anda paragraph-iv filings a complete review
DESCRIPTION
anda filingTRANSCRIPT

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
96
ANDA PARAGRAPH-IV FILINGS: A COMPLETE REVIEW
Useni Reddy Mallu*, Sekhar Karanam and Panyala Srinath ReddyDept. of Chemistry, Sri Krishnadevaraya University, Anantapur, Andhra Pradesh, India
INTRODUCTION
INTRODUCTION
ISSN:2249-5347IJSID
International Journal of Science Innovations and Discoveries An International peerReview Journal for Science
Review Article Available online through www.ijsidonline.info
Received: 16.07.2012
Accepted: 29.08.2012
*Corresponding Author
Address:
Name:Dr. Useni Reddy MalluPlace:Dept. of Chemistry,Sri KrishnadevarayaUniversity, Anantapur,Andhra Pradesh, IndiaE-mail:[email protected]
ABSTRACTThe present review throws light on the criticalities and complexities involved in the paragraphIV ANDA filings with USFDA and the intellectual property litigations evolving around between thebranded manufacturers and generic companies. Hatch-Waxman Act grant generic manufacturers theability to mount a validity challenge without incurring the cost of entry or risking enormous damagesflowing from any possible infringement. The entry of generic product in to the market allows the public tohave access to low cost medicines equivalent to brand products. Once multiple generic firms enter themarket, prices fall, often dramatically. The current generic approval process involves submission of onlyan abbreviated new drug application (ANDA) that relies on FDA approval of the pioneer drug (or patentedbrand name drug) to demonstrate safety and effectiveness. The generic drug does not require proof that itis safe and effective--that has already been proven by the brand name drug manufacturer and throughmany years of clinical use in the general population. Generally, the ANDA has to establish only that thegeneric drug is bioequivalent to the pioneer drug (i.e., the reference-listed drug for which therapeuticequivalence is sought). The Hatch-Waxman Act gave additional protection to the inventors of newdrugs,both by lengthening patent terms and by providing guaranteed periods of data exclusivity.Inexchange, Hatch-Waxman made it easier for generic drug manufacturers to enter the market with a copyof the drug, either by waiting until the patent expires or by challenging weak patents. To encouragegeneric manufacturers to identify and challenge weak patents, Hatch-Waxman offered a sort of incentiveto the generic challenger. The first generic manufacturer to file for approval with the FDA, as paragraph IVis entitled to 180 days of “generic exclusivity” when it first enters the market. During that period, othergeneric drug makers are prohibited from entering the market. The point was to offer an incentive,encouraging generic firms to challenge and invalidate bad patents (or invent around them) early andoften, and accordingly get generic drugs on to the market earlier. Multiple ANDA filings on the same daywould lead to sharing the exclusivity between generic manufacturers. 30-month FDA stay period andpatent infringement litigations became a common scenario in the growing global generic pharmaceuticalindustry. with very little downside and huge upside, exclusivity is the driving force to the huge increase infirst-to-file Paragraph IV filings.Keywords: USFDA-ANDA submission, Orange book, Paragraph-IV filing, 30months stay period,Litigations, 180days exclusivity, Multiple ANDA submission, Hatch Waxman act, CFR , First to file andcase studies.

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
97
INTRODUCTIONGeneric drugs are important options that allow greater access to health care for public. They are copies of brand-namedrugs and are the same as those brand name drugs in dosage form, safety, strength, route of administration, quality,performance characteristics and intended use. Health care professionals and consumers can be assured that FDA approvedgeneric drug products have met the same rigid standards as the innovator drug. All generic drugs approved by FDA have thesame high quality, strength, purity and stability as brand-name drugs. And, the generic manufacturing, packaging, and testingsites must pass the same quality standards as those of brand name drugs.The Food and Drug Administration (FDA or USFDA) is an agency of the United States Department of Health andHuman Services, one of the United States federal executive departments. The FDA is responsible for protecting and promotingpublic health through the regulation and supervision of food safety, tobacco products, dietary supplements, prescription andover-the-counter pharmaceutical drugs (medications), vaccines, biopharmaceuticals, blood transfusions, medical devices,electromagnetic radiation emitting devices (ERED), and veterinary products. The FDA also enforces other laws, notablySection 361 of the Public Health Service Act and associated regulations, many of which are not directly related to food ordrugs. These include sanitation requirements on interstate travel and control of disease on products ranging from certainhousehold pets to sperm donation for assisted reproduction.The FDA is led by the Commissioner of Food and Drugs, appointed by the President with the advice and consent of theSenate. The Commissioner reports to the Secretary of Health and Human Services. The 21st and current Commissioner is Dr.Margaret A. Hamburg. She has served as Commissioner since February 2009.USFDA have several offices and centers for monitoring the different aspects. The details are represented in figure-1.
Figure No-1: USFDA centers and offices.FDA's Center for Drug Evaluation and Research (CDER) regulates over-the-counter and brand name and genericprescription drugs. In this function, CDER supports innovation and plays a key role in helping to advance new drugdevelopment. CDER is the division for monitoring the pharmaceuticals, and manages the current good manufacturing practice(cGMP) regulations for pharmaceutical manufacturing and drug approvals. FDA has Legal Authorities, for saving human healthand providing less cost medicines and cosmetics and even life threatening diseases also. The list of authorities, acts andamendments are tabulated in table-1.

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
98
Table No-1: List of FDA regulations and amendmentsYear Regulation or Amendment1902 Biologics Control Act1906 Pure Food and Drug Act1938 Federal Food, Drug and Cosmetics Act1944 Public Health Service Act1951 Food Drug and Cosmetics Act Amendments1962 Food Drug and Cosmetics Act Amendments1966 Fair Packaging and Labeling Act1976 Medical Device Regulation Act1984 Drug Price Competition –Patent Term Restoration Act (Hatch Waxman Act)1987 Prescription Drug Marketing Act1988 Antidrug Abuse Act1990 Nutrition Labeling and Eduction Act1992 Prescription Drug User Fee Act1994 Dietary Supplement Health and Education Act1997 Food Drug Modernization Act2002 Bioterrorism Act2002 Medical Device User Fee and Modernization Act (MDUFMA)2003 Animal Drug User Fee Act2007 Food and Drug Administration Amendments Act of 2007
Code of Federal Regulations (CFR)The CFR is published by federal government of the United States. CFR is the codification of the general and permanentrules and regulations. CFR can be called as administrative law. The CFR is divided into 50 titles that represent broad areassubject to Federal regulation as listed below.CFR, Title 21 governs the food and drugs within the US for the FDA, the Drug Enforcement Administration (DEA), andthe Office of National Drug Control Policy (ONDCP). 21 CFR is divided into three chapters: Chapter I ,deals the FDA, Chapter IIdeals the Drug Enforcement Administration and Chapter III deals Office of National Drug Control Policy. Each chapter againdivided in to parts. Part- 314 deals the applications for FDA approval to market a new drug from patents to ANDA submission.In this ,part 314 again divided into subparts like subpart A, B, C, D, E, F, G, H and I.Title 21 CFR Part 11 of the Code of Federal Regulations deals with the FDA guidelines on electronic records andelectronic signatures in the US. Part 11, as it is commonly called, defines the criteria under which electronic records andelectronic signatures are considered to be trustworthy, reliable and equivalent to paper records In general, Part 11 insistsdrug makers, medical device manufacturers, biotech companies, biologics developers, contract research organizations (CRO)and other FDA-regulated industries, with some specific exceptions, to implement controls, including audits, systemvalidations, audit trails, electronic signatures, and documentation for software and systems involved in processing electronicdata that are (a) required to be maintained by the FDA predicate rules or (b) used to demonstrate compliance to a predicaterule. A predicate rule is any requirement set forth in the Federal Food, Drug and Cosmetic Act (FDC Act), the Public HealthService Act, or any FDA regulation other than Part 11.The FDA will approve the new drug products (NDA) by evaluating the safety and efficacy. The innovator shouldprovide the scientific and clinical data, must usually from at least two well-controlled clinical studies. Once approved the drugproduct, FDA will assure the market protection with the forms of exclusivity which is a creation of law. Exclusivity enables thedrug product to have exclusive, or monopoly, status in the market for a certain number of years (five years for a new chemical
Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
99
entity (NCE) and other periods of time for different situations.) Exclusivity means that the FDA cannot legally approve ageneric drug application for that product until the exclusivity period expires.Table No-2: List of CFR titles
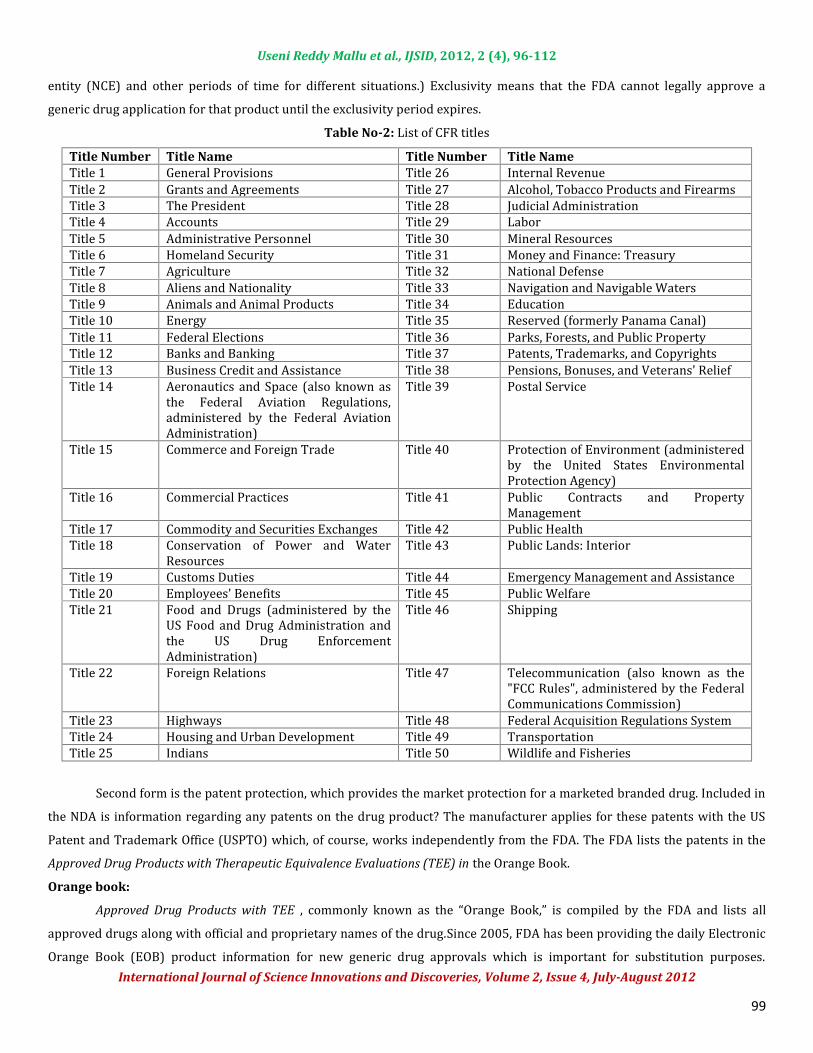
Title Number Title Name Title Number Title NameTitle 1 General Provisions Title 26 Internal RevenueTitle 2 Grants and Agreements Title 27 Alcohol, Tobacco Products and FirearmsTitle 3 The President Title 28 Judicial AdministrationTitle 4 Accounts Title 29 LaborTitle 5 Administrative Personnel Title 30 Mineral ResourcesTitle 6 Homeland Security Title 31 Money and Finance: TreasuryTitle 7 Agriculture Title 32 National DefenseTitle 8 Aliens and Nationality Title 33 Navigation and Navigable WatersTitle 9 Animals and Animal Products Title 34 EducationTitle 10 Energy Title 35 Reserved (formerly Panama Canal)Title 11 Federal Elections Title 36 Parks, Forests, and Public PropertyTitle 12 Banks and Banking Title 37 Patents, Trademarks, and CopyrightsTitle 13 Business Credit and Assistance Title 38 Pensions, Bonuses, and Veterans' ReliefTitle 14 Aeronautics and Space (also known asthe Federal Aviation Regulations,administered by the Federal AviationAdministration)Title 39 Postal Service
Title 15 Commerce and Foreign Trade Title 40 Protection of Environment (administeredby the United States EnvironmentalProtection Agency)Title 16 Commercial Practices Title 41 Public Contracts and PropertyManagementTitle 17 Commodity and Securities Exchanges Title 42 Public HealthTitle 18 Conservation of Power and WaterResources Title 43 Public Lands: InteriorTitle 19 Customs Duties Title 44 Emergency Management and AssistanceTitle 20 Employees' Benefits Title 45 Public WelfareTitle 21 Food and Drugs (administered by theUS Food and Drug Administration andthe US Drug EnforcementAdministration)Title 46 Shipping
Title 22 Foreign Relations Title 47 Telecommunication (also known as the"FCC Rules", administered by the FederalCommunications Commission)Title 23 Highways Title 48 Federal Acquisition Regulations SystemTitle 24 Housing and Urban Development Title 49 TransportationTitle 25 Indians Title 50 Wildlife and FisheriesSecond form is the patent protection, which provides the market protection for a marketed branded drug. Included inthe NDA is information regarding any patents on the drug product? The manufacturer applies for these patents with the USPatent and Trademark Office (USPTO) which, of course, works independently from the FDA. The FDA lists the patents in the
Approved Drug Products with Therapeutic Equivalence Evaluations (TEE) in the Orange Book.Orange book:
Approved Drug Products with TEE , commonly known as the “Orange Book,” is compiled by the FDA and lists allapproved drugs along with official and proprietary names of the drug.Since 2005, FDA has been providing the daily ElectronicOrange Book (EOB) product information for new generic drug approvals which is important for substitution purposes.

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
100
Previously, a first-time-generic product approved early in the month would not be published in the EOB for several weeks. Themonthly EOB update goal is by the end of the following month’s second work week (e.g., January’s EOB will be updated by theend of the second full work week in February).The EOB content includes NDA approvals in the EOB month they were approved. NDA application numbers arepreceded with “N”; ANDA or Generic product as of the date of the daily update. Generic application numbers are preceded with“A”; All product changes received and processed as of the monthly update date and Discontinued products will be processed asof the date of publication. There will be circumstances where a product is discontinued in one month, however, it will bereported in a different month's EOB. For example, the Orange Book Staff received a letter November 7 that the product hasbeen discontinued from manufacturing and marketing. The Orange Book subsequently updates the October EOB on November14. The product will show in the October EOB that it is discontinued even though the date of discontinuance is the day that theOrange Book Staff receives notification (November 7)). Patent information, also updated daily in the EOB, as of the date of thedaily update. You can download the orange book annual edition in PDF form from the FDA site.FDA Requirements for Generic Drugs
Same active ingredients, same dosage form, same labeled strength, labeling as the brand-name product. Generic drug manufacturers must show that a generic drug is bioequivalent to the brand-name drug, which means thegeneric version delivers the same amount of active ingredients into a patient's bloodstream in the same amount of time asthe brand-name drug. Generic drug manufacturers must fully document the generic drug's chemistry, manufacturing steps, and quality controlmeasures. Firms must assure the FDA that the raw materials and finished product meet specifications of the U.S. Pharmacopoeia, theorganization that sets standards for drug purity in the United States. Firms must show that a generic drug will remain potent and unchanged until the expiration date on the label. Firms must comply with federal regulations for good manufacturing practices and provide the FDA a full description offacilities they use to manufacture, process, test, package, and label the drug. The FDA inspects manufacturing facilities toensure complianceHATCH WAXMAN (HW) Act:The Drug Price Competition and Patent Term Restoration Act of 1984, usually refered to as the Hatch-Waxman Act,was designed to promote generics while leaving intact a financial incentive for research and development. It allows generics towin FDA marketting approval by submitting bioequivelence studies (as opposed to clinical data, which is costlier to compile).It also grants a period of additional marketing exclusivity to make up for the time a patented pipeline drug remains indevelopment. This extension cannot exceed five years, and it is in addition to the 20 years exclusivity granted by the issuanceof a patent. Additionally, the generic manufacturer must file a certification regarding patents listed in the Orange Book (alsoknown as Approved Drug Products with Therapeutic Equivalence Evaluations). A paragraph IV certification states that thepatent is invalid or will not be infringed and begins a process by which that question may be answered by the courts prior toexpiration of the patent. Under Hatch-Waxman, FDA approval of an ANDA is automatically stayed for 30 months when a patentowner files a patent infringement lawsuit within 45 days of receiving a paragraph IV notification. During the stay, the FDA isprohibited from approving another ANDA. Additionally, the first ANDA is granted a 180-day exclusivity period, as an incentive

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
101
whereby the generic company does not have competition from other generic companies and can both establish market shareand charge a higher price.Although the net effect of HW on pharmaceutical innovation is ambiguous, its effect on generic drug development hasbeen explicit, and the effect on consumers has been beneficial. Hatch-Waxman resulted in increased ANDA applications andparagraph IV (P-IV)challenges, especially since 1998. There has also been a high success rate for patent invalidation,particularly formulation and polymorph patents. Since Hatch-Waxman, virtually all top-selling drugs not covered by patentface generic competition; whereas pre–Hatch-Waxman, only 35% had generics available. Similarly, today more than 70% ofprescriptions are for generics.An unexpected consequence resulting from HW, especially over the past 10 years, has been the filing of patentapplications by generic companies and increased generic research and development for branded products. Table-3 representsthe major loopholes in HW act.Table No-3: Major loopholes in HW act
Major Loopholes of Hatch Waxman Act1 Sharing of 180 day exclusivity by several generic manufacturers2 Filing “Citizen petitions” to freeze the ANDA application3 Risky generic launch strategies4 No prohibition against “authorized generics”HATCH WAXMAN (HW) Amendments:HW was amended several times to close these loopholes and/or decrease generic drug approval times. Additionally,over the years, the FDA issued many guidance documents clarifying Hatch- Waxman with the goals of reducing genericapproval time; improving ANDA application quality to avoid multiple review cycles; avoiding time-consuming legal delays; andclosing “loopholes” which delayed competition. Unfortunately, anticompetitive strategies and loopholes continue today andfuel the momentum for further legislative Hatch-Waxman reform. HW act amendments were tabulated in table-4.
Table No-4: HW act amendmentsAmendments of Hatch Waxman Act1 Medicare prescription drug, Improvement and Modernization Act of 2003(MMA)2 FDA Amendments act of 20073 QI programme supplemental funding Act of 2008
ANDA submission and Approval Process:An Abbreviated New Drug Application (ANDA) contains data which when submitted to FDA's Center for DrugEvaluation and Research, Office of Generic Drugs, provides for the review and ultimate approval of a generic drug product.Once approved, an applicant may manufacture and market the generic drug product to provide a safe, effective, low costalternative to the public.When patent information is submitted for a new drug application the patent information is included in the OrangeBook. Through the abbreviated new drug application (“ANDA”) process, a party may obtain FDA approval of generic drugswithout clinical trials if the drug is a bioequivalent of a drug previously granted NDA approval. ANDA approval requires that anapplicant make a patent certification “with respect to each patent issued by the United States Patent and Trademark Office

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
102
that, in the opinion of the applicant and to the best of its knowledge, claims the reference listed drug or claims a use of suchlisted drug for which the applicant is seeking approval Certification requires the ANDA applicant to state that: (1) the NDAholder submitted no patent to the FDA; (2) any patent submitted has expired; (3) the date the applicable patent expires; or (4)that “the patent is invalid, unenforceable, or will not be infringed by the manufacture, use, or sale of the drug product forwhich the abbreviated application is submitted.”Patent ExclusivityANDA has four types of submissions, Paragraph-I (P-I): patent information relating to the innovator drug has not beenfiled; Paragraph-II (P-II): the patent has expired; Paragraph-III (P-III) : ANDA will be approved after the particular patentexpires and Paragraph-IV (P-IV): Challenging a particular patent that is listed by the Innovator in the "Orange Book."Paragraph I, II and III:A certification under P-I or II permits the ANDA to be approved immediately when otherwise eligible. A certificationunder P-III indicates that the ANDA may be approved on the patent expiration date. A certification under P-III indicates thatthe ANDA may be finally approved when the patent expires.
Figure No-2: Types of Paragraph filings for ANDA submission.Paragraph IV (P-IV) CertificationThe basis of P-IV certification is The Drug Price Competition and Patent Term Restoration Act that came to be knownas the “Hatch-Waxman Act.”The Hatch-Waxman rules created processes and incentives for both branded and generic companies involving challenges topatents. Branded pharmaceutical companies are required to list patents involving composition of matter (substance), formulation,and method of use in the Food and Drug Administration “s (FDA) Orange Book. When applying to enter the market with a generic form of a reference product, the generic company files an AbbreviatedNew Drug Application (ANDA) and certifies against patents listed in the Orange Book. The certification states that itsgeneric product does not infringe on the listed patents or that those patents are not enforceable, called a Paragraph IVfiling.

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
103
If the generic company files an ANDA with a Paragraph IV certification, then the branded company is notified within 20days. After the notice, the branded company has 45 days to file a patent infringement action against the generic company.After the suit has been filed, the FDA cannot approve of the application until the generic company successfully defends thesuit or until 30 months, whichever comes first.“30 MONTH STAY” PERIODIn patent litigations between branded pharmaceutical companies and generic manufacturers, where the genericcompany files a P-IV certified ANDA and is attempting to sell a product that is equivalent to the branded company’s product,much of the litigation focus and drama revolves around the so-called “30-month” FDA stay date. Under the HW Act, once thebranded pharmaceutical company files a timely complaint, the FDA will not grant the generic company final approval of itsproduct for “30-months,” absent a court decision that the patent is not infringed, invalid or unenforceable.
Figure No-3: Paragraph-IV submission and 30months stay
eCTDsubmissionthrough ESGFDAAcceptanceLetter
USFDA-ANDA Paragraph-IV applicant
SubmissionRejection SubmissionRejection
CTDsubmission(Hard copy)
ANDA applicant raises the letter to innovator byexplaining existing patent is invalid
30months stayis effectiveSuit on ANDA
No 30months stay ,FDA will give theapproval
Innovator can file a patent infringementlawsuit, even if authentic claim by the ANDAapplicantNo Suit on ANDA

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
104
In principle, the 30-month FDA stay period is designed to provide the court enough time to resolve the brandedpharmaceutical company’s patent issues, and the FDA sufficient time to review the generic company’s ANDA. Recognizing thatdelay in the litigation by either party could have serious implications, the HW Act states that a court may either shorten orlengthen the 30-month period if “either party to the action failed to reasonably cooperate in expediting the action.” Becausethe generic company would like to enter the market sooner than the 30-month FDA stay period, it must invoke the statute totry to shorten the stay period, alleging that the branded company has improperly delayed the litigation The submission of an ANDA for a drug product application containing a P- IV certification may be sued for patentinfringement. If the NDA holder or patent owner files a patent infringement suit against the ANDA applicant within 45days of the receipt of notice, FDA could not give final approval to the ANDA for at least 30 months from the date of thatnotice. This 30-month stay will delay approval of the generic drug product unless the court reaches a decision earlier in thepatent infringement case or otherwise orders a longer or shorter period for the stay. A court may modify the length of astay, under the FD&C Act, “if either party in the action failed to reasonably cooperate in expediting the action.” (21 U.S.C.335(j)(5)(iii)) Under FDA’s traditional interpretation of the HW Amendments, multiple 30-month stays have been possible. Submissionof newly issued patents after an ANDA application has been filed with FDA has required the appropriate certification andnotice to the NDA holder and patent owner with the possibility of a 30-month stay if patent infringement litigationresulted. As a result, there have been a number of instances in which delays in ANDA approval have exceeded 30-months. A recent review of FDA’s records indicates that out of 100 ANDA applications 4 are filed under paragraph IV certificationswith patent challenges. However, we note that a significant number of these products have high dollar value annual sales. Once 30 months stay period have passed, the FDA can approve the ANDA application, though litigation is ongoing and theapproved generic firm can market their product based on the favorable court decisions.Strategies adopted by generics and brands on grounds of HW Loopholes1. One such strategy is the “at risk” product launch by generic companies which basically involves launching theproduct prior to a district court clearance on litigations.2. Another strategy involves filing an ANDA ,for the sake of filing and having little chance for success. Under HW, thefirst ANDA filer, regardless of the outcome of the subsequent patent infringement litigation, is entitled to the 180-day exclusivity. For patents found valid, upon expiration, a new ANDA need not be filed. While the intent was toprovide other generic companies from benefiting when they had risked nothing, the actual result is the maximumnumber of P-IV filings on day one.3. Yet another strategy is the market entry of “authorized generics” (“AG”), which are brand companies’ own“generics” marketed either through brand product relabeling (perhaps through a subsidiary) or licensingagreements with another company. The company entitled to 180- day exclusivity loses revenue, even if the“authorized generic” is the result of a P-IV litigation settlement agreement between itself and the brandmanufacturer.4. The brand company benefits from AG sales even after expiration of the brand drug patent. Marketing an AG duringthe 180- day exclusivity period violates the first ANDA’s statutorily granted “exclusivity,” ie, the policy and intent of

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
105
HW, and may run afoul of the Sherman Act or State Unfair Competition Acts. Moreover, critics of AG argue thatpackaging and labeling the branded drug as a generic are tantamount to “misbranding.”5. The generic history reveals that, the generic companies must choose to either incur the cost and risk of continuingpatent litigation and face fewer returns during the 180-day period, or settle. Via a “reverse payment agreement” abrand company pays a generic company a substantial amount of money to either not file an ANDA application orstay out of the market for a specified period of time. In some cases, the amount is actually greater than the genericcompany would actually make if it were to market the generic.6. While under the MMA the 180-day exclusivity is forfeited for generic applications entering anticompetitiveagreements as determined by the Federal Trade Commission (FTC) or attorney general or failing to enter themarket within 75 days of effective approval or 30 months after submission of an application entitling the 180 days,most often the forfeiture is avoided by legal maneuvers and “bottlenecks” continue. Legislation to prohibit “reversepayment” agreements and “bottleneck” strategies has been unsuccessful many times.7. The FDA has no expertise or resources with which to resolve complex questions of patent coverage. Generic andbrand companies may only resolve any patent disputes in private litigation. Patent “evergreening”—that is,obtaining additional patents on specific features of a drug product, eg, isomers, polymorphs, metabolites,intermediates, process patents, or double patenting— became a popular practice among brand companies withexpiring Orange Book patents to extend the monopoly on a drug. Multiple 30-month stays were triggered when theNDA holder added new patents to the Orange Book, whereas each patent required a separate paragraph IVcertification, thereby resulting in a new lawsuit hence, new 30-month stays. In some cases, companies had chainsof patents which were added to the Orange Book in a staggered manner to keep generics out of the market. Underthe medicate modernization act (MMA), generic companies must now only certify to Orange Book patents listed atthe time of initial ANDA filing, effectively preventing multiple 30-month stays.8. A Citizen Petition is a request that anyone can make to the FDA regarding a specific aspect of a pending application.Questioning the quality of a competitor’s product through last minute Citizen Petitions with dubious or frivolousclaims is yet another method to delay generic competition. Additionally, the Citizen Petition results in a “hold” onthe ANDA until decided.9. Although statutorily the FDA commissioner may take up to 90 days to investigate the petition, it often takes muchlonger. Generic manufacturers would like to prohibit or limit the filing of Citizen Petitions by innovatormanufacturers.Multiple ANDA’S:The related issues of “shared exclusivity”” arise in the following situation1) Multiple generic applicants submit ANDAs with paragraph IV certifications to the same patent(s) on the same first day;The agency explained that when, on the same day, more than one applicant submits an ANDA for the same drugcontaining a paragraph IV certification to a listed patent, and no such certification was submitted previously, all the applicantswill share exclusivity. Exclusivity will be triggered for all first applicants for a specific listed patent when one of them begins tomarket its product (or on the date of any court decision finding that patent invalid, unenforceable, or not infringed, if earlier).

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
106
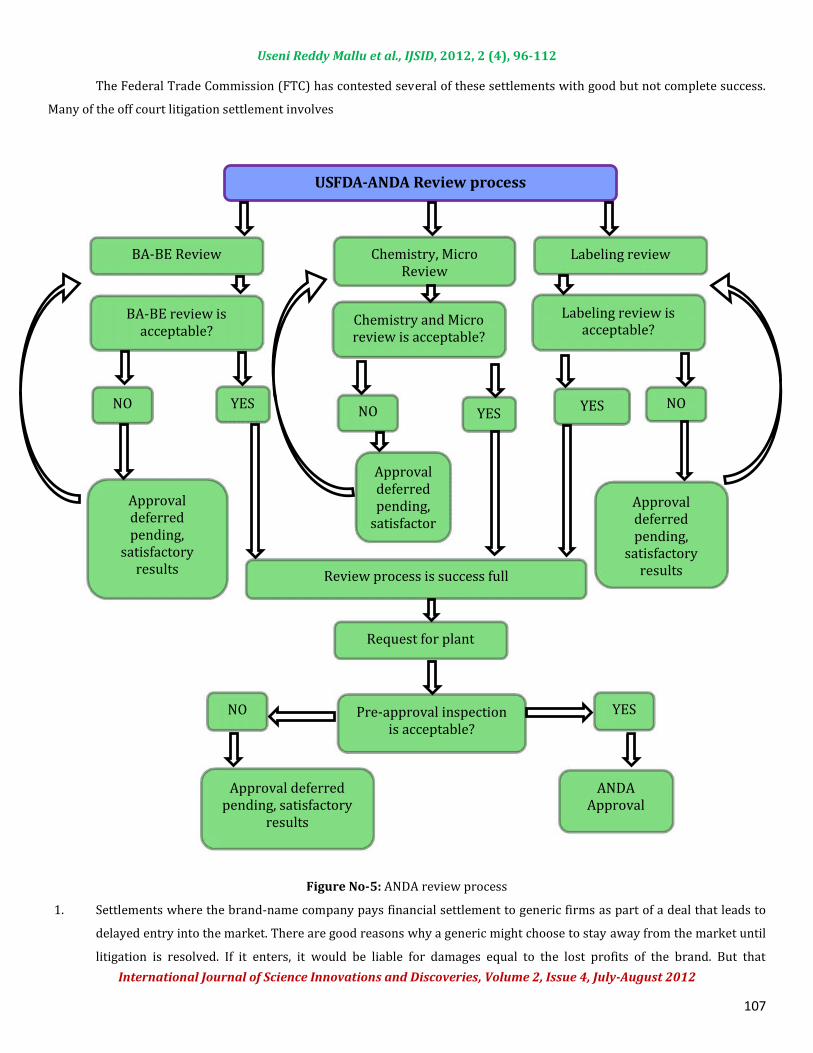
Figure No-4: Multiple ANDA submission with Paragraph-IV certificationANDA Review ProcessANDA review can be proceeded in different parts like BABE review, Chemistry and micro review and Labeling reviewprocedures. Drug substance critical review points are API characterization, Synthesis, In-process methods, Specifications andstability studies. Drug product critical review points are formulation development, QbD approach, manufacturing process, In-process controls, Analytical methods for in-process and finished product, specification limits and stability.Litigation & SettlementsParagraph IV of the HW Act provides a mechanism for the litigation of pharmaceutical patent infringement disputes.Many of these cases have been settled with reverse payments by the brand to the generic in return for delayed generic productentry by using the available loop holes of HW Act.
Paragraph IV ANDA Submission
Generic Companies A & B filing on Day 1 Generic Companies C & D filing on Day 2Within 20 days’ notice by generic firmsto brand manufacturer about patentinvalidationWithin 45 days’ brand manufacturer willsue the generic applicant
If not suedFDA canreview andapprove theANDAIf sued “30 month Stay period” for FDAapproval begins
FDA extend final approval afterexpiry of “30 months stay” for firstapplicants A & BAfter final approval Generics A & Breceives 180 day exclusivity and canstart marketing based on litigationstatus
Generics C & D receives finalapproval only after completingthe 180 day exclusivity byeither generic firm A or B
Applicant-A and B areeligible for 180days
and C and D areconsidered for normalapproval (no 180days)
Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
107
The Federal Trade Commission (FTC) has contested several of these settlements with good but not complete success.Many of the off court litigation settlement involves
Figure No-5: ANDA review process1. Settlements where the brand-name company pays financial settlement to generic firms as part of a deal that leads todelayed entry into the market. There are good reasons why a generic might choose to stay away from the market untillitigation is resolved. If it enters, it would be liable for damages equal to the lost profits of the brand. But that
USFDA-ANDA Review process
BA-BE Review Chemistry, MicroReview
Request for plantinspection
Labeling reviewBA-BE review isacceptable? Chemistry and Microreview is acceptable?
Review process is success full
Pre-approval inspectionis acceptable?Approval deferredpending, satisfactoryresults ANDAApproval
NOYES NONO YESYES
NO
Labeling review isacceptable?
Approvaldeferredpending,satisfactoryresultsApprovaldeferredpending,satisfactory results Approvaldeferredpending,satisfactoryresults
YES

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
108
stipulation does not require allowing for payments to the generic. There are also good reasons why a brand mightrequest a stay against a generic's entry, especially if it feels that it has a good case and the generic is unlikely to havethe money to pay a damage claim The reason is that the payment structure can create an incentive for the generic todrag out the litigation, particularly if either (1) it is likely that it will lose, or (2) there is a second generic who willlikely be able to enter after 180 days, regardless of when the litigation is resolved.2. But the pure cash deals would lead to an antitrust problem, and to overcome that some firms have attempted thefollowing structure. The generic agrees for delayed entry, the brand-name company provides the entrant with cash,and the generic entrant provides the brand manufacturer with some alternate considerations like licensing to patentsthat the generics hold. This was the basic structure of the Schering-Upshur-Smith deal that the FTC ultimatelychallenged.3. Some deals have involved Royalty Payments like receiving licenses to enter the market, either immediately or on adelayed basis, with the generic entrant paying royalties to the brand4. One final device that has been used to game the system involves the licensing of pseudo-generics (Authorizedgenerics) in settlements. A pseudo-generic is the branded drug sold as a generic under license from the brand.5. However the Federal Trade Commission monitors these sort of gamings and if found antitrust, sues both the partieswith charging them as anticompetitive behavior in market.180 Days Market ‘EXCLUSIVITY”HW Act provides an incentive of 180 days market exclusivity to the “first” ANDA applicant who challenges a listedpatent by filing a paragraph IV certification and thereby runs the risk of having to defend a patent infringement suit. AfterUSFDA approval and successful court litigations, begins commercial marketing of the generic drug product with 180 daysmarket exclusivity. In some cases, a generic manufacturer who obtains 180-day exclusivity may be the sole marketercompeting with the brand product for market shareIn some cases, if there is no court decision and the first applicant does not begin commercial marketing of the genericdrug, there may be prolonged or indefinite delays in the beginning of the first applicant’s 180-day exclusivity period. Until aneligible ANDA applicant’s 180-day exclusivity period has expired, FDA cannot approve subsequently submitted ANDAs for thesame drug. This is possible even if the later ANDAs are otherwise ready with tentative approval and the sponsors are willing tobegin marketing immediately. Therefore, an ANDA applicant who is eligible for exclusivity can often delay other genericcompetition for the brand product by delaying the market entry.Only a “first” ANDA applicant filing a paragraph IV certification shall be eligible for exclusivity. If an applicant changesfrom a paragraph IV certification to a paragraph III certification, for example, upon losing its patent infringement litigation, theANDA will no longer be eligible for exclusivity.The 180-day exclusivity provision has been the subject of considerable litigation and administrative review in recentyears, as the courts, industry, and FDA have sought to interpret it in a way that is consistent both with the statutory text andwith the legislative goals underlying the HW Amendments. A series of Federal court decisions beginning with the 1998 Movacase describe acceptable interpretations of the 180-day exclusivity provision, identifiying potential problems in implementingthe statute, and establish certain principles to be used by the Agency in interpreting the statute. As described in a June 1998guidance for industry, FDA currently is addressing on a case-by-case basis those 180-day exclusivity issues not addressed byexisting regulations.

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
109
One of the most basic changes to the 180-day exclusivity program, in view of the legal challenges to FDA’s regulations,is the determination by the courts of the meaning of the phrase “court decision.” The courts have determined that the “courtdecision” that can begin the running of the 180-day exclusivity period may be the decision of the district court, if it finds thatthe patent at issue is invalid, unenforceable, or will not be infringed by the generic drug product. FDA had previouslyinterpreted the “court decision” that could begin the running of 180-day exclusivity (and the approval of the ANDA) as the finaldecision of a court from which no appeal can be or has been taken - generally a decision of the Federal Circuit. FDA’sinterpretation had meant that an ANDA applicant could wait until the appeals court had finally resolved the patentinfringement or validity question before beginning the marketing of the generic drug.FDA had taken this position so that the generic manufacturer would not have to run the risk of being subject topotential treble damages for marketing the drug, if the appeals court ruled in favor of the patent holder. The currentinterpretation means that if the 180-day exclusivity is triggered by a decision favorable to the ANDA applicant in the districtcourt, the ANDA sponsor who begins to market during that exclusivity period now may run the risk of treble damages if thedistrict court decision is reversed on appeal to the Federal Circuit. As a practical matter, it means that many generic applicantsmay choose not to market the generic and thus the 180-day exclusivity period could run during the pendency of an appeal.Recent P-IV Litigations- Case Studies
1. ANDA litigation over Lupin's generic version of FORTAMET (metformin extended-release tablets). Sciele Pharma v.
Lupin, No. 2012-1228 (Fed. Cir.)-Presently the case under litigation in district court
2. Genzyme and against ANDA applicants Roxane, Sandoz, and Anchen in the paragraph IV litigation concerningHECTOROL (doxercalciferol),- District court decision was in favour of the brand company and concluded that the ANDA
holders had infringed the claims in OB patent and can go for appeal in Federal court if required.3. Cephalon in connection with its ANDA for a generic version of Cephalon's FENTORA (fentanyl citrate) buccal tablets.Cephalon v. Watson-In the court decision Cephalon could not prove infringement of patents by Watson for two patents
,but Watson failed to prove invalidity of Cephalon patent 6,264,981 and declared a permanent injuction against Watson
till the expiry of this patent in October,2019.
4. Generic version of sumatriptan/naproxen sodium that competes with Treximet(R) (sumatriptan and naproxensodium) sold by GlaxoSmithKline in patent infringement lawsuit against Par, Alphapharm Pty Ltd., TevaPharmaceuticals USA, Inc., and Dr. Reddy's Laboratories, Inc - The District Court ruled U.S. Patent Nos. 6,060,499 (the‘499 patent) and 6,586,458 (the ‘458 patent) to be valid, enforceable and infringed by Par Pharmaceutical, Inc. (Par),Alphapharm Pty Ltd. (Alphapharm), and Dr. Reddy’s Laboratories, Inc. (DRL). A third patent, U.S. Patent No. 7,332,183(the ‘183 patent) covering the Treximet formulation was held to be valid, enforceable and infringed by Par and DRL. The‘183 patent was not asserted against Alphapharm.
5. Atorvastatin litigations,Pfizer Vs Ranbaxy;Ranbaxy will Market Generic Atorvastatin in the U.S. with 180 DaysExclusivity from Nov. 30, 2011 - RANBAXY AND PFIZER SETTLE LIPITOR LITIGATION WORLDWIDE,Under the termsof the agreement, Ranbaxy will have a license to sell generic versions of Atorvastatin and the fixed-dose combination ofAtorvastatin-Amlodipine besylate in the United States effective Nov. 30, 2011.
6. Mylan's 10 mg and 20 mg omeprazole delayed-release capsules, which are the generic versions of AstraZeneca LP'sPrilosec ;Mylan wins Omeprazole patent litigation
7. AstraZeneca’s Crestor(Rosuvastatin Calcium tablets) Patent litigation, case against Teva Pharmaceuticals, Mylan Labs,Par Pharmaceutical, Aurobindo Pharma, Sun Pharmaceutical-Litigations are in progress and all the generic firms got
tentative approval

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
110
180 Days Exclusivity-Forfeiture Provisions
Forfeiture Events(I) Failure to Market(II) Withdrawal of Application(III) Amendment of Certification(IV) Failure to Obtain Tentative Approval within 30 Months(V) Agreement with another Applicant, the Listed Drug Application Holder, or a Patent Owner(VI) Expiration of All PatentsFDA Practice• “It is FDA’s practice to make decisions on eligibility for 180-day exclusivity only in the context of specificANDAs those are otherwise eligible for approval.”• When FDA must make an approval decision for an ANDA, it will inform the applicant that it is(1) A first applicant and entitled to exclusivity(2) A first applicant that has forfeited its exclusivity(3) Eligible only for tentative approval (TA) due to another’s exclusivity(4) Eligible for final approval (FA) because another forfeited exclusivity• FDA will consider whether there has been forfeiture when approval of a subsequent ANDA maybe blocked by a first applicant’s exclusivity.Failure to MarketThe first applicant fails to market the drug by later of (aa) the earlier of 75 days after final approval; or 30 monthsafter ANDA submission; or The date that is 75 days after the date as of which, as to each of the patents that qualified the FA forexclusivity, at least one of the following has occurred:
Final decision of invalidity or non-infringement. Settlement order entering final judgment. NDA holder delists patent from Orange Book. Withdrawal of Application “The first applicant withdraws the application or the Secretary considers the application to have been withdrawn as aresult of a determination by the Secretary that the application does not meet the requirements for approval underparagraph (4).” Administrative withdrawal due to poor quality ANDA?
Amendment of Certification
o “The first applicant amends or withdraws theo certification for all of the patents with respect too which that applicant submitted a certificationo qualifying the applicant for the 180-day exclusivityo 21 CFR 314.94(a)(12)(viii)(A) requires amendment ofo a p.IV to a p.III after a final judgment of infringement

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
111
Pseudo Generics:To prevent generic completion from increasing their market sharing, some brand-name forms produce genericsthemselves called pseudo-generics. Literature and practical experiences are provide strong evidence that brand-name drugproduct prices are decreasing drastically after entering the generic drug product in to the market. Brand-name companiesalways have an incentive to purchase its competitor. By developing pseudo-generics product brand-name Company canincrease its market value.CONCLUSIONGreater access to generic drugs will reduce health care costs because the price of generic drugs is typically muchlower than the brand-name drug. Reducing expensive lawsuits over drug patents and making the approval process moreefficient will also help to lower national health care costs by reducing the cost of bringing safe and effective generic drugs tomarket. Throughout the years since enactment of Hatch-Waxman, the US patent and regulatory systems continued to evolve.The average drug research and development time is now 10 years, and the total costs often exceed $750 million. At 5 years,the United States currently has the shortest data exclusivity protection for innovative drugs.. The Hatch-Waxman Act didencourage growth of the generic industry and provided brand companies with incentives. The incentives led to gaming of thesystem, however. While some maintain that the FTC has adequate authority to challenge litigation settlements that may beanticompetitive, others believe the judicial system is not the appropriate venue to resolve these issues.. Clearly, there is a needto promote an “innovation culture,” not a “litigation culture.” Looking back at history of the Hatch-Waxman Act, its successesand failures, and looking forward at proposed legislative reforms, it is clear that the pharmaceutical scale must remainbalanced.DISCLAIMERThe purpose of this review article is solely educational. This review article is built from authors work and experience.REFERENCES
CFR:1. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm/2. http://www.fda.gov/medicaldevices/deviceregulationandguidance/databases/ucm135680.htm3. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=312.30Pseudo Generics:4. Aidan Hollis, 2003. "The Anti-Competitive Effects of Brand-Controlled "Pseudo- Generics" in the Canadian PharmaceuticalMarket, "Canadian Public Policy, University of Toronto Press, vol. 29(1), pages 21-31.5. Jorge Mestre Ferrándiz, 1999. "The impact of generic goods in the pharmaceutical industry, "Health Economics, John Wiley& Sons, Ltd., vol. 8(7), pages 599-612.6. FerrÆndiz, J. The impact of generic goods in the pharmaceutical industry. Health Economics 1999; 8: 599-612.7. Kong, Y. and Seldon, J. Pseudo-generic products and barriers to entry in pharmaceutical markets. Review of IndustrialOrganization 2004; 25: 71-86.180DAYS:8. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079342.pdf9. http://www.ftc.gov/be/v990016.shtm

Useni Reddy Mallu et al., IJSID, 2012, 2 (4), 96-112
International Journal of Science Innovations and Discoveries, Volume 2, Issue 4, July-August 2012
112
10. http://www.orangebookblog.com/files/180day_exclusivity_forfeiture.pdf11. http://aspe.hhs.gov/mits/text/titleXI/1102.html12. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072851.pdf13. http://www.cov.com/files/Publication/1d6a8448-b41b-4909-a385-3363886a80cd/Presentation/PublicationAttachment/a13273a4-65a3-498f-9a59-371017d71f42/761.pdfParagraph-IV:14. http://thomsonreuters.com/content/science/pdf/ls/newport_paragraphIV_webinar_slides.pdf15. http://leehu.myweb.uga.edu/papers/lee_hu_jmp.pdfHW litigation:16. http://www.orangebookblog.com/hatchwaxman_litigation/17. Weiswasser, E.S. and S.D. Danzis. 2003. \The Hatch-Waxman Act: History, Structure, and Legacy." An-titrust Law Journal71:585.18. Wiggins, S.N. and R. Maness. 2004. \Price competition in pharmaceuticals: the case of anti-infectives."Economic Inquiry42(2):247{263.19. Scott Morton, F.M. 1999. \Entry Decisions in the Generic Pharmaceutical Industry." The RAND Journal of Economics30(3):421{440.20. Scott Morton, F.M. 2000. \Barriers to entry, brand advertising, and generic entry in the US pharmaceutical industry."International Journal of Industrial Organization 18:1085{1104.21. Derzko, N.M. 2005. \The Impact of Recent Reforms of the Hatch-Waxman Scheme on Orange Book Strategic Behavior andPharmaceutical Innovation." IDEA 45(2):165{265.22. DiMasi, J.A., R.W. Hansen and H.G. Grabowski. 2003. \The price of innovation: new estimates of drug development costs."Journal of Health Economics 22(2):151{185.23. Chen, T. 2007. \Authorized Generics: A Prescription for Hatch-Waxman Reform." Virginia Law Review 93(2):459{513.24. http://www.mcguirewoods.com/news-resources/publications/commercial_litigation/ANDA_patent_issues.pdf25. Terry G. Mahn, The Hatch-Waxman Act During Patent Prosecution and Beyond, Food and Drug Law J. (1999)26. http://www.pm360online.com/f4_0712_Bio_Pharma_medicine_healthcare_patent_expiration_Pfizer_180_Day_War_for_Lipitor27. http://regulatory.usc.edu/Articles/LabelingConsiderations.pdf28. http://www.regulatoryone.com/2012/01/anda.html