analysis of the glucocerebrosidase (gba) and gtp cyclohydrolase-1 (gch1) genes in a uk cohort of...
TRANSCRIPT

Analysis of the Glucocerebrosidase (GBA) and GTP Cyclohydrolase-1 (GCH1) genes in a UK cohort of familial Parkinson’s Disease
patients.
Piers Fulton, West Midlands Regional Genetics Laboratory
Trainee Project 2009/2010

Parkinson’s Disease (PD)
Progressive, incurable neurodegenerative disorder.
Affects >1% of 55 year olds, >3% of over 75 year olds.
Second most common neurodegenerative disorder after Alzheimer’s Disease.
Clinical Symptoms:
Bradykinesia – slowed movement.Rest tremor.
Muscle rigidity.
Dementia (20%), hallucinations, postural instability, ‘freezing’/motor blocks, dysautomnia, dystonic cramps.
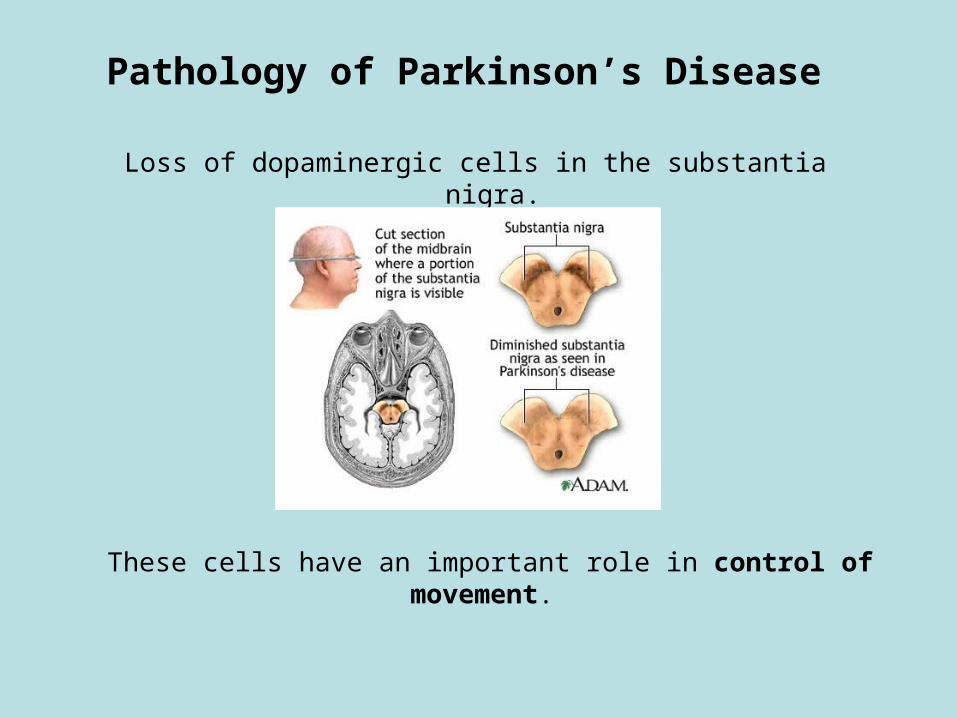
Pathology of Parkinson’s Disease
Loss of dopaminergic cells in the substantia nigra.
These cells have an important role in control of movement.

Causes of PD
Vast majority of cases: no strong family history or clear cause of PD (idiopathic).
Multifactorial disease. Genetics and environmental risk factors (age, sex, pesticide exposure, head injury) play a role.
Small proportion of cases (<5%): Mendelian inheritance pattern.

Mendelian Causes of PD
Genetests lists seven genes or mapped loci with an established link to AD or AR PD.
Most relevant to this project:
LRRK2.
Mutations cause 2-7% of Autosomal Dominant familial PD.
LRRK2 PD follows a similar disease course to idiopathic PD.

This study
1. Test a UK-recruited cohort of familial Parkinson’s Disease patients for mutations in the PD candidate genes GBA and
GCH1.
2. Identify any patients carrying mutations in LRRK2 and GBA/GCH1.

Study Cohort
1. 46 UK-based familial PD patients recruited at the Queen Elizabeth Hospital, Birmingham.
2. All unrelated, each has at least 2 affected family members, apparent AD inheritance.
3. LRRK2 previously sequenced in all patients: 7 carriers of pathogenic mutations.
4. GCH1 previously sequenced in one patient: GCH1 point mutation identified.
Patient recruitment, LRRK2 and GCH1 sequencing: David Nicholl, Alistair Lewthwaite and colleagues.

Glucocerebrosidase (GBA)
1q21
Essential for breakdown of glucosylceramide in lysosomes.
Mutations in GBA may cause Gaucher disease.
AR lysosomal storage disorder.
Parkinsonism is a rare complication.

Glucocerebrosidase (GBA)
Previously reported association between GBA mutations,
increased PD risk, lower age of PD onset in patients unaffected by Gaucher disease.
Limited previous research into frequency of GBA mutations in familial PD.
Nichols et al (2009): 12.8% of large US familial PD cohort carried one of five pathogenic GBA mutations, v. 5.3% of controls.

Sequencing of all 11 exons + flanking regions of GBA.
GBA has a 98% homologous pseudogene (GBAP) 16kb downstream, complicates primer design.
~10kb Long Range-PCR fragment amplified, nested PCR and sequencing of exons from diluted LR-PCR product.
GBA locus should be amplified even if partly replaced with
GBAP-derived sequence by homologous recombination/gene conversion (~12% of mutant alleles are recombinant).
Sequencing GBA

Complete sequence obtained for 45/46 patients.
Five potentially pathogenic missense variants identified in five
patients.
GBA sequencing results
Patient HGVS
1 p.Arg159Trp/N
2 p.Gly234Glu/N
3 p.Arg502Cys/N
4 p.Thr408Met/N
5 p.Leu483Pro/
p.Thr408Met
Severe (null): p.Arg159Trp, p.Gly234Glu.
Medium/severe: p.Leu483Pro.
Mild: p.Arg502Cys.
Mild/polymorphism: p.Thr408Met.

Patient 4: p.Thr408Met/NAlso carries a LRRK2 frameshift mutation.
Patient 5: p.Leu483Pro/p.Thr408Met
p.Thr408Met is unlikely to be a major contributor to the PD phenotype.
Inheritance pattern (AD) not consistent with inheritance of two
disease-causing alleles.
No family history of Gaucher disease in 5’s family.
(Conclusion consistent with Eblan et al. 2006, Gan-Or et al. 2009).
Is p.Thr408Met/N pathogenic?

4/45 patients (8.9%) carry a pathogenic GBA mutation.
Mean age of onset of GBA mutation carriers = 42.3 years (Range: 28-54) .
Mean age of GBA mutation non-carriers = 59.4 years (Range: 30-78).
P-value (T-test):
p=0.0098.
GBA mutation carriers have a significantly earlier age of onset than non-carriers.
Do GBA mutations alter age of onset in fPD?

14q21.1-q22.2
Essential role in dopamine synthesis.
Mutations in GCH1 can cause Dopa-Responsive Dystonia (DRD).
AD neurological disorder, variable penetrance, usually childhood
onset.
Parkinsonism is a common symptom of DRD.
GTP Cyclohydrolase-1 (GCH1)

Family investigated at Queen Elizabeth Hospital:
Variable neurological phenotype: PD (2 patients) or DRD (two
patients).
Novel GCH1 missense mutation (p.Glu2Gly) identified in DRD and PD patients, not in unaffected family members.
Conclusion: family carries a GCH1 mutation with variable
phenotype.
My project: screen GCH1 in fPD cohort, which includes the proband from the previously investigated family.
Why test GCH1?

1. Sequencing
PCR, sequencing of all six coding exons and flanking regions.
2. MLPA
P052-C1 Parkinson probemix, contains probes for 5 GCH1 exons (exon 4 not covered).
Testing GCH1

1. Sequencing
p.Glu2Gly confirmed in original proband.
No other known pathogenic mutations identified.
UV, p.Pro23Leu identified in patient 19. Also carries LRRK2 p.Gly2019Ser.
Same GCH1/LRRK2 genotype in patient 19’s affected sister.
Results

Is p.Pro23Leu pathogenic?
Previously identified in DRD patients and controls: evidence suggests either a mild mutation or a polymorphism.
Inheritance pattern (AD) not consistent with inheritance of two
disease-causing alleles.
Further work to assess co-segregation with disease in patient 19’s family is justified.
Results

2. MLPA
Poor quality results due to low DNA concentration and quality.
No GCH1 deletions or duplications detected in 39/46 patients (7 patients failed).
Results

GBA
Pathogenic GBA mutations detected in 8.9% of a UK-recruited cohort of unrelated familial PD patients.
GBA mutation carriers found to have a significantly lower age of
PD onset than non-carriers.
GBA mutations are a risk factor in familial PD in the UK population.
Conclusions

GCH1
No known pathogenic GCH1 mutations identified.
One unclassified variant identified in a LRRK2 mutation carrier.
GCH1 mutations are not a significant risk factor in fPD in the UK population.
Conclusions

Inheritance of mutations in multiple genes
No conclusive evidence of an additive effect of multiple risk alleles.
LRRK2 mutation/GBA UV and LRRK2 mutation/GCH1 UV combination each seen in 1 patient.
Conclusions

Queen Elizabeth Hospital, Birmingham:David Nicholl, Alistair Lewthwaite.
West Midlands Regional Genetics Laboratory:Fiona Macdonald, Jennie Bell, Yvonne Wallis, Matthew Smith,
Kirsten McKay, Hayley Simm and sequencing team.
Acknowledgements