an adaptive mutation in ns2 is essential for efficient production of infectious 1b/2a chimeric...
TRANSCRIPT

Virology 422 (2012) 224–234
Contents lists available at SciVerse ScienceDirect
Virology
j ourna l homepage: www.e lsev ie r .com/ locate /yv i ro
An adaptive mutation in NS2 is essential for efficient production of infectious 1b/2achimeric hepatitis C virus in cell culture
Katie Chan ⁎, Guofeng Cheng, Rudolf K.F. Beran, Huiling Yang, Todd C. Appleby, Maria V. Pokrovskii,Hongmei Mo, Weidong Zhong, William E. Delaney IVGilead Sciences, Inc., 333 Lakeside Drive, Foster City, CA 94404, USA
⁎ Corresponding author. Fax: +1 650 522 5890.E-mail address: [email protected] (K. Chan).
0042-6822/$ – see front matter © 2011 Elsevier Inc. Alldoi:10.1016/j.virol.2011.10.022
a b s t r a c t
a r t i c l e i n f oArticle history:Received 5 August 2011Returned to author for revision24 August 2011Accepted 22 October 2011Available online 17 November 2011
Keywords:Cell culture adaptationHepatitis C virusChimeraAdaptive mutationsCon1JFH1Virus production
The development of JFH1 based intergenotypic recombinants which exploit the unique replication character-istics of JFH1 has made it possible to study infectious HCV encoding the structural genes of additional HCVgenotypes including genotype 1b. Although, intergenotypic 1b/2a chimeric genomes replicate efficiently intransfected cells they produce very low viral titers, limiting the utility of this system. Here, intergenotypic1b/2a variants were generated by serially passaging the virus in a novel highly permissive Huh-7 cellclone. The adapted virus was 1000-fold more infectious than the parental unadapted virus and six adaptedmutations were identified throughout the genome. Of the mutations identified, L839S in the NS2 gene wasthe most critical for the adapted phenotype by enhancing the infectivity of assembled viral particles. Overall,the efficient production of infectious 1b/2a virus particles will facilitate the discovery and characterization ofinhibitors targeting steps that involve the structural genes of genotype 1b HCV.
© 2011 Elsevier Inc. All rights reserved.
Introduction
As many as 170 million people worldwide are chronically-infectedwith hepatitis C virus (HCV) and consequently are at risk of developingchronic hepatitis, cirrhosis and hepatocellular carcinoma (WHO, 1999).HCV is a positive strandRNA virus that belongs to the Flaviviridae family(Bartenschlager and Pietschmann, 2005). The HCV genome is approxi-mately 9600 nucleotides encoding a single open reading frame thatencodes about 3000 amino acid residues (Miyamoto et al., 2006). Theresulting polyprotein is cleaved by host and virally-encoded proteasesinto ten distinct proteins, which fall into two families: 1) structuralproteins (Core, E1, E2) and 2) non-structural proteins (p7, NS2, NS3,NS4A, NS4B, NS5A, and NS5B).
One hallmark of HCV is its large genetic diversity. HCV is classifiedinto six major genotypes (designated 1–6) and several subtypes(WHO, 1999). Genotype 1 (particularly 1a and 1b) is the most preva-lent HCV genotype in North America, Europe and Asia. The currentstandard of care for chronic HCV infection is combination therapywith pegylated interferon-α and ribavirin. Unfortunately, patientswith genotype 1 HCV are the least responsive to this treatment (lessthan 50%), making the current standard of care unsatisfactory formany patients (Pawlotsky et al., 2007). In contrast, response rates
rights reserved.
for genotypes 2 and 3 are significantly higher (~80%) (Mangia andAndriulli, 2008). The difference in response rates may be, in part,due to differences in viral characteristics between genotypes(Miyamoto et al., 2006). Further research is required to better under-stand the molecular mechanisms underlying such differences; suchan understanding may ultimately translate into more effective thera-pies for HCV.
In the past, HCV research has been hampered by the lack of viralcell culture systems that allow for the study of the complete virallife cycle (Lanford et al., 2009). A major breakthrough occurred in2005 when Wakita et al. (2005) demonstrated that the genotype 2avirus isolate JFH1 could replicate to high levels inside cells in theabsence of cell culture adaptive mutations, and most importantlyproduce infectious virus particles in cell culture (HCVcc). However,to date, no other isolates, and notably no genotype 1 strains, havebeen identified that support robust replication and production ofinfectious virus particles in cell culture.
To overcome this limitation, several groups have constructed viablechimeric genomes that comprised JFH1 non-structural genes and struc-tural genes from heterologous HCV strains (Gottwein et al., 2007, 2009;Pietschmann et al., 2006; Scheel et al., 2008; Yi et al., 2007; Zhang et al.,2008). This has made it possible to study infectious HCV encoding thestructural genes of additional HCV genotypes, including genotype 1b(Con1). Intergenotypic Con1/JFH1 chimeric genomes maintain robustRNA replication and allow production of infectious particles with struc-tural proteins derived from genotype 1b. However, viral titers produced

Fig. 1. Improved permissiveness of 497-5 Lunet cells to HCVcc infection and viralspread. A) Improved permissiveness of 497-5 Lunet cells to HCVcc infection. Huh-7Lunet, Lunet-CD81 and 497-5 Lunet cells were infected with Jc-1 Rluc virus at variousMOIs ranging from 0.00036 to 0.36. Luciferase activity, a marker for active viral replica-tion, was measured 72 h post-infection and is indicated as relative light units. All datapoints were generated from at least two independent experiments and expressed asmean±SEM. B) Improved HCVcc viral spread after transfection in 497-5 Lunet cells.Huh-7 Lunet, Lunet-CD81, and 497-5 Lunet cells were transfected with in vitro tran-scribed Jc-1 Rluc RNA. Virus spread was measured by NS5A immunostaining to deter-mine the percentage of HCV-positive cells at days 1, 2, 4, and 7 post-transfection.All data points were generated from two separate experiments and expressed asmean±SEM.
225K. Chan et al. / Virology 422 (2012) 224–234
by intergenotypic chimeras are typically low, which poses challengesformany cell-based applications. To produce high virus titers, two com-ponents appear to be required: 1) a host cell that supports both efficientviral infection and RNA replication and 2) a virus that is able to efficient-ly replicate, assemble and be released by the host cells; such virusestypically contain cell culture adaptive mutations (Bartenschlager andPietschmann, 2005; Woerz et al., 2009).
To obtain higher infectious Con1/JFH1 chimeric genomes, we firstidentified a novel Huh-7 Lunet subclone that was more permissiveto viral infection and spread. This cell clone was named 497-5 Lunet.Second, the Con1/JFH1 genomewas constructed and passaged seriallyover 497-5 Lunet cells, to generate adapted Con1/JFH1 virus variantscapable of producing more infectious virus particles. Eventually, anadapted virus was selected that produced infectious viral titers1000-fold higher than the parental strain. Adaptive mutations wereidentified in both the structural and non-structural regions of theadapted virus; however mutations in the non-structural regionshad the greatest impact on the enhanced infection phenotype. Usingreverse genetics, mutations were analyzed to determine their impacton virus production and spread. A panel of known HCV inhibitorswith distinct mechanisms of actionwas also tested to validate the util-ity of the adapted virus for antiviral screening. Overall, the generationof this novel adapted Con1/JFH1 HCV chimera is expected to facilitatethe identification and characterization of novel HCV inhibitors includ-ing those that target steps in virus entry, assembly or release of geno-type 1b HCV.
Results
Identification of a novel Huh-7 Lunet subclone highly permissive toHCVcc infection
Huh-7 Lunet cells support robust HCV replication, but are limitedin their permissiveness to HCV infection and spread (Koutsoudakiset al., 2007). This phenotype has been linked to low levels of CD81,a key HCV entry factor on the surface of Huh-7 Lunet cells,(Koutsoudakis et al., 2007). We previously transduced Huh-7 Lunetcells with human CD81 to generate the cell line Lunet-CD81(Pokrovskii et al., 2011). This cell line had improved susceptibility toHCV infection but had reduced transfection efficiency compared toparental Huh-7 Lunet cells. To identify novel Huh-7 Lunet subclonesthat were more permissive for HCV infection, we screened a panel of77 stable replicon cell lines for CD81 surface expression by FACS. The77 individual replicon cell lines were derived from drug resistanceselections with various HCV inhibitors and had each been expandedfrom a single colony. A Huh-7 Lunet clone that naturally expressedhigher levels of CD81 was identified (termed 497-5 Lunet) and wasthen cured of HCV RNA by treatment with IFN-α and two otheranti-HCV compounds (BILN-2061 and BMS-790052).
The permissiveness of 497-5 Lunet cells to HCV infection wascompared to Huh-7 Lunet and Lunet-CD81 cells by infection with aJc-1 Rluc virus at various MOIs. Luciferase activity was used as amarker for viral replication and was measured 72 h post-infection.As shown in Fig. 1A, Jc-1 Rluc viral infection was most efficient in497-5 Lunet cells, as the luciferase activity was highest in these cellsat all MOIs>0.001. The ability of 497-5 Lunet cells to support in-creased viral spread was also assessed. In vitro transcribed Jc-1 Rlucviral RNA was transfected into 497-5 Lunet, Lunet-CD81 and Huh-7Lunet cells, and viral spread in each cell line was monitored byNS5A immunostaining over 7 days (Fig. 1B). Viral spread was highestin 497-5 cells, as the percentage of NS5A positive cells increased from3% on day 1 to 86% on day 7 post-transfection. In comparison, Lunet-CD81 cells only achieved 46% NS5A positive cells, while the parentalHuh-7 Lunet cells only achieved 23% NS5A positive cells on day 7.Together, these findings suggest that viral spread and infection ismuch more efficient in 497-5 Lunet cells compared to Lunet-CD81
and the parental Huh-7 Lunet cell line. Because 497-5 Lunet cellssupport robust HCV replication after transfection and enhancedviral spread, they were used in the subsequent studies to developCon1/JFH1 cell culture adapted viruses.
Generation of cell-culture adapted 1b/2a chimeric virus
A genotype 1b/2a Con1/JFH1 chimeric HCV genome was con-structed as described by Pietschmann et al. (2006). In this construct,the core, E1, E2, p7 and NS2 genes of Con1 were fused with NS2 andthe remaining non-structural genes of JFH1 at a junction site locatedbetween the first and second putative transmembrane domains ofNS2. In vitro transcribed Con1/JFH1 RNA was transfected into 497-5Lunet cells and the cells were maintained by serial passaging at a1:3 dilution every 2 to 3 days. At each passage, cells were immuno-stained for NS5A to monitor the degree of infection and supernatantswere collected to determine viral titer (Fig. 2). Initially, viral titersproduced after transfection were very low, reaching a maximum of102 TCID50/ml; titers did not increase for 17 days. Immunostainingfor NS5A indicated 20% positive cells at day 2 post transfection andthis increased to almost 40% at day 4, likely reflecting a combinationof translation from input RNA and then translation from newly syn-thesized RNA produced by intracellular HCV replication. Since viraltiters produced were initially low, the percentage of infected cellsafter the first passage subsequently decreased, reaching as low as10% NS5A positive cells at day 17. A sharp rise in infection was
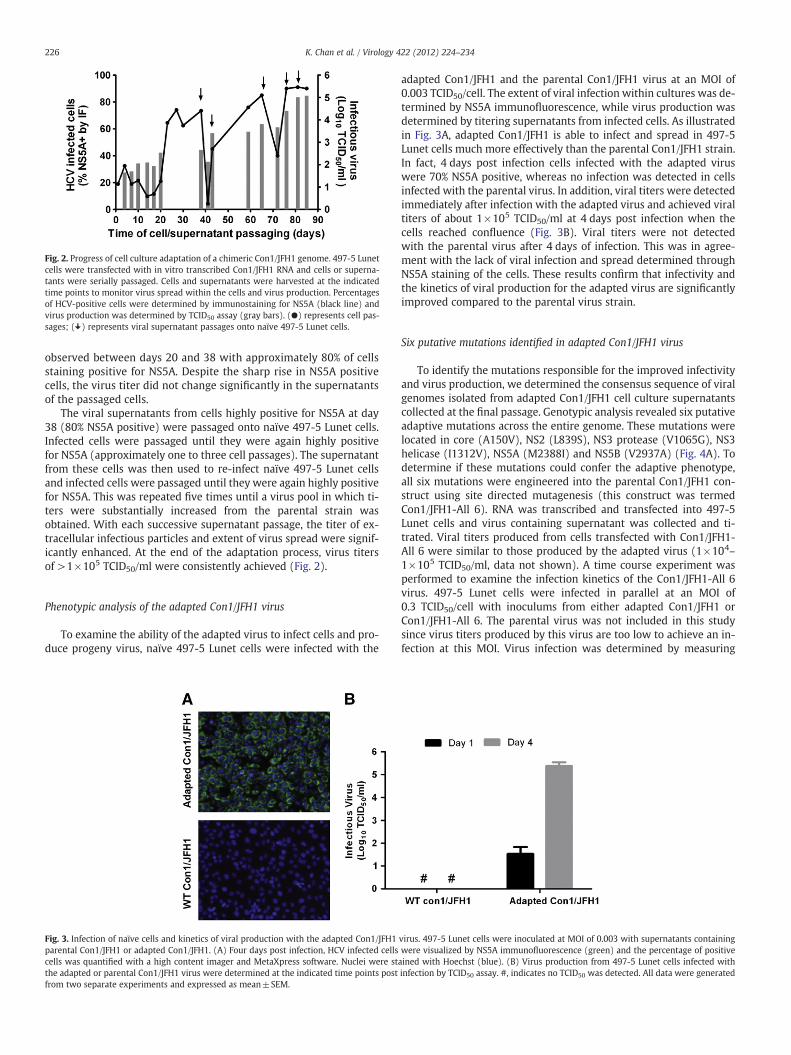
Fig. 2. Progress of cell culture adaptation of a chimeric Con1/JFH1 genome. 497-5 Lunetcells were transfected with in vitro transcribed Con1/JFH1 RNA and cells or superna-tants were serially passaged. Cells and supernatants were harvested at the indicatedtime points to monitor virus spread within the cells and virus production. Percentagesof HCV-positive cells were determined by immunostaining for NS5A (black line) andvirus production was determined by TCID50 assay (gray bars). (●) represents cell pas-sages; ( ) represents viral supernatant passages onto naïve 497-5 Lunet cells.
226 K. Chan et al. / Virology 422 (2012) 224–234
observed between days 20 and 38 with approximately 80% of cellsstaining positive for NS5A. Despite the sharp rise in NS5A positivecells, the virus titer did not change significantly in the supernatantsof the passaged cells.
The viral supernatants from cells highly positive for NS5A at day38 (80% NS5A positive) were passaged onto naïve 497-5 Lunet cells.Infected cells were passaged until they were again highly positivefor NS5A (approximately one to three cell passages). The supernatantfrom these cells was then used to re-infect naïve 497-5 Lunet cellsand infected cells were passaged until they were again highly positivefor NS5A. This was repeated five times until a virus pool in which ti-ters were substantially increased from the parental strain wasobtained. With each successive supernatant passage, the titer of ex-tracellular infectious particles and extent of virus spread were signif-icantly enhanced. At the end of the adaptation process, virus titersof >1×105 TCID50/ml were consistently achieved (Fig. 2).
Phenotypic analysis of the adapted Con1/JFH1 virus
To examine the ability of the adapted virus to infect cells and pro-duce progeny virus, naïve 497-5 Lunet cells were infected with the
Fig. 3. Infection of naïve cells and kinetics of viral production with the adapted Con1/JFH1parental Con1/JFH1 or adapted Con1/JFH1. (A) Four days post infection, HCV infected cellscells was quantified with a high content imager and MetaXpress software. Nuclei were stthe adapted or parental Con1/JFH1 virus were determined at the indicated time points postfrom two separate experiments and expressed as mean±SEM.
adapted Con1/JFH1 and the parental Con1/JFH1 virus at an MOI of0.003 TCID50/cell. The extent of viral infection within cultures was de-termined by NS5A immunofluorescence, while virus production wasdetermined by titering supernatants from infected cells. As illustratedin Fig. 3A, adapted Con1/JFH1 is able to infect and spread in 497-5Lunet cells much more effectively than the parental Con1/JFH1 strain.In fact, 4 days post infection cells infected with the adapted viruswere 70% NS5A positive, whereas no infection was detected in cellsinfected with the parental virus. In addition, viral titers were detectedimmediately after infection with the adapted virus and achieved viraltiters of about 1×105 TCID50/ml at 4 days post infection when thecells reached confluence (Fig. 3B). Viral titers were not detectedwith the parental virus after 4 days of infection. This was in agree-ment with the lack of viral infection and spread determined throughNS5A staining of the cells. These results confirm that infectivity andthe kinetics of viral production for the adapted virus are significantlyimproved compared to the parental virus strain.
Six putative mutations identified in adapted Con1/JFH1 virus
To identify the mutations responsible for the improved infectivityand virus production, we determined the consensus sequence of viralgenomes isolated from adapted Con1/JFH1 cell culture supernatantscollected at the final passage. Genotypic analysis revealed six putativeadaptive mutations across the entire genome. These mutations werelocated in core (A150V), NS2 (L839S), NS3 protease (V1065G), NS3helicase (I1312V), NS5A (M2388I) and NS5B (V2937A) (Fig. 4A). Todetermine if these mutations could confer the adaptive phenotype,all six mutations were engineered into the parental Con1/JFH1 con-struct using site directed mutagenesis (this construct was termedCon1/JFH1-All 6). RNA was transcribed and transfected into 497-5Lunet cells and virus containing supernatant was collected and ti-trated. Viral titers produced from cells transfected with Con1/JFH1-All 6 were similar to those produced by the adapted virus (1×104–
1×105 TCID50/ml, data not shown). A time course experiment wasperformed to examine the infection kinetics of the Con1/JFH1-All 6virus. 497-5 Lunet cells were infected in parallel at an MOI of0.3 TCID50/cell with inoculums from either adapted Con1/JFH1 orCon1/JFH1-All 6. The parental virus was not included in this studysince virus titers produced by this virus are too low to achieve an in-fection at this MOI. Virus infection was determined by measuring
virus. 497-5 Lunet cells were inoculated at MOI of 0.003 with supernatants containingwere visualized by NS5A immunofluorescence (green) and the percentage of positiveained with Hoechst (blue). (B) Virus production from 497-5 Lunet cells infected withinfection by TCID50 assay. #, indicates no TCID50 was detected. All data were generated

Fig. 4. Six mutations identified in the adapted virus confer the adapted phenotype. Genotypic analysis of cell culture adapted Con1/JFH1 virus identified six putative adaptive mu-tations. (A) Schematic map of Con1/JFH1 chimeric genome, Con1 (gray) and JFH1 (white). Locations of the six adaptive mutations found are indicted. (B) A time course of intra-cellular NS3–4A protease activity (as a marker for active viral replication) was performed on 497-5 Lunet cells infected with either the adapted virus or a Con1/JFH1 virusencoding the six putative adaptive mutations “All 6”. (C) Percentages of HCV-positive cells were determined by immunostaining for NS5A at days 2 and 4 of the infection timecourse. All data were generated from three separate experiments and expressed as mean±SEM.
227K. Chan et al. / Virology 422 (2012) 224–234
intracellular NS3–4A protease activity, which serves as a marker forviral RNA replication and protein production. The infection kineticsof the Con1/JFH1-All 6 virus was indistinguishable from that of thecell culture adapted virus (Fig. 4B). In addition, the percentage ofcells infected with HCV was determined by NS5A immunofluores-cence at day 2 and day 4 of the infection. The percentage of cellsinfected by either the All 6 or adapted virus was comparable at bothtime points (Fig. 4C). These results indicate that the six mutationsidentified were sufficient to confer tissue culture adaptation.
The NS2 L839S mutation is the main determinant for enhancing Con1/JFH1viral infectivity and production
To identify individual mutations that are essential for producingthe adapted phenotype, each mutation was reverted back to itswild-type residue in Con1/JFH1-All 6 or inserted individually intothe parental wild-type construct. 497-5 Lunet cells were transfectedwith transcribed mutant Con1/JFH1 RNAs and virus spread and pro-duction were monitored in transfected cells for 7 days.
Reversion of the NS2 mutation (L839S) had the most significantimpact on virus spread (Fig. 5A). Without this mutation, the extentof virus spread was reduced to wild-type levels. Similarly, reversion
of the NS2 mutation had the most profound impact on virus produc-tion, reducing viral titers 100-fold below those of the Con1/JFH1-All 6virus at day 7 (Fig. 5B). Reversion of the other mutants generally hadlittle impact on viral spread and infectious titer by the end of theseven day experiment. However, some of the mutation reversions,particularly I1312V (NS3 helicase) and V2937A (NS5B), appeared toreduce the kinetics of viral infection and secretion as evidenced bythe fewer NS5A positive cells and lower titers at earlier time points.These data suggest that the NS2 mutation L839S is the major contrib-utor to the enhanced phenotype while the other mutations are likelyto play an incremental role in the adaptation.
Consistent with observations made with the individual revertants,only the NS2 mutation L839S significantly enhanced virus spread as asingle mutant engineered in the wild-type Con1/JFH1 virus (Fig. 5C);this mutation increased the percentage of infected cells from 16%(WT) to 75% (NS2 L839S) at day 7 post transfection (Fig. 5C). Allother single mutations did not enhance virus spread above wildtype levels. Similarly, virus titers were increased 100-fold comparedto wild type levels when the single mutation in NS2 was present(Fig. 5D), however this mutation alone was not sufficient to repro-duce the viral titers achieved by the adapted virus. Constructs withindividual mutations in NS3 helicase (I1312V), NS5A (M2388I) and

Fig. 5. Contribution of individual mutations to enhanced virus spread and production. Six mutations were individually reverted to wild-type in the Con1/JFH1-All 6 construct or insertedindividually in thewild-type Con1/JFH1 parental clone. 497-5 Lunet cells were transfectedwith in vitro transcribed RNA and the percentage of HCV infected cells was visualized by NS5Aimmunofluorescence over a seven day period. Viral titers were also determined by TCID50 assay for supernatants collected over a seven day period. The effects of reverting one of the sixmutations in the adapted virus on virus spread (A) or virus production (B) in transfected cells are indicated. The effects of adding singlemutations to thewild-type parental virus on virusspread (C) or virus production (D) in transfected cells are indicated. All data were generated from at least two separate experiments and expressed as mean±SEM.
228 K. Chan et al. / Virology 422 (2012) 224–234
NS5B (V2937A) had no impact on virus production, producing viraltiters similar to wild-type. Interestingly, the mutations in core(A150V) and NS3 protease (V1065G) impaired virus production rela-tive to the wild type, as no viral titers were detected over the sevenday period (Fig. 5D). These data indicate that no single mutationwas sufficient to confer the full adapted phenotype.
Overall, data gathered from mutation introduction and reversionexperiments converged to suggest that the NS2 mutation (L839S) isthe primary adaptive mutation, as it had the most profound effect inenhancing virus production and spread and was the most cripplingwhen absent.
The NS2 L839S mutation enhances the production of competentinfectious particles
Since the NS2 L839S mutation was the most critical for enhancingviral spread and production of infectious titer, subsequent analyseswere focused on dissecting the role of this mutation in the adaptivephenotype. To determine whether enhanced RNA replication was in-volved in the adapted phenotype, we measured RNA levels followingelectroporation of the mutants into Huh-7 Lunet cells. Huh-7 Lunetcells were selected for these experiments since they limit virus spreaddue to low level expression of CD81 on their cell surfaces (Kaul et al.,2007). For this reason, HCV RNA levels and virus titers measured inHuh-7 Lunet cells will originate almost exclusively from cells thattake up HCV RNA during the electroporation procedure, rather thanfrom cells infected by progeny virus following the first round ofviral replication. As shown in Fig. 6A, the levels of intracellular RNAin transfected cells were comparable between Con1/JFH1 wild-typeand the All 6 construct. Similarly, the NS2 single mutant (L839S) or
NS2 revertant (Rev L839S) also replicated at levels comparable towild-type. Therefore, the enhanced virus production seen by theadapted virus was not due to increased intracellular RNA replication.HCV RNA levels in the supernatants of each transfected culture werealso similar (Fig. 6B), despite the substantial differences found inthe extracellular viral titers between each construct (Fig. 6D). Thisfinding suggests that the adaptive phenotype cannot be explainedby an increase in HCV RNA replication or release of HCV genomes,since the L839S or the other adaptive mutations had no apparenteffect on these steps in the viral life cycle.
To investigate if improved viral titers were due to either enhancedassembly or release of infectious particles, we measured the amountof infectious virus present within cells and also in the supernatantsafter transfection. The wild-type, All 6, NS2 (L839S) and ReversedNS2 (Rev L839S) RNAs were electroporated into Huh-7 Lunet cellsand viral titers were measured in intracellular (Fig. 6C) and extracel-lular (Fig. 6D) compartments. Higher viral titers were measuredwhen the NS2 mutation was present (All 6 and NS2 [L839S] virusesvs. the wild-type and Reversed NS2 [Rev L839S] viruses) indicatingthat this mutation facilitated the formation or accumulation of infec-tious virus. The increases in intracellular viral titers were reflectedin the increases in the viral titers released to the supernatants forthe NS2 mutant containing viruses. In addition, reversion of the NS2mutation from the All 6 virus reduced both intracellular and extracel-lular viral titers to levels comparable to the wild-type virus bothintracellularly and extracellularly, again suggesting that the NS2mutation was most critical for the enhanced accumulation of infec-tious viral particles during tissue culture adaptation.
To confirm that increased titers were due to increased formation oraccumulation of infectious virus particles, we determined intracellular

Fig. 6. NS2 (L839S) mutation enhances specific infectivity and efficiency of assembled particles. RNA transcripts were transfected into Huh-7 Lunet cells. At days 2, 4, and 7 posttransfection, cells were harvested and supernatants were collected for quantification of HCV RNA levels by qRT PCR and infectivity by TCID50 assay. Virus producing cells werewashed and lysed by repetitive cycles of freeze and thaw to determine intracellular HCV RNA levels and infectivity. (A) Intracellular HCV RNA levels; (B) intracellular virus titersand (C) intracellular specific infectivity, were determined. In parallel, (D) extracellular HCV RNA levels; (E) extracellular virus titers and (F) extracellular specific infectivity weredetermined in supernatants. All data were generated from at least two separate experiments and expressed as mean±SEM.
229K. Chan et al. / Virology 422 (2012) 224–234
and extracellular specific infectivities (Figs. 6E and F). Specificinfectivity was calculated as the ratio of infectious titers to HCVRNA content within each compartment. As shown, only the presenceof the NS2 mutation was required to achieve the high specific infec-tivity, as All 6 and the NS2 single mutant showed similar specificinfectivity values. When the NS2 mutation was absent (Rev L839S),the specific infectivity reverted back to wild-type levels. Thus, theadaptive mutation in NS2 alone substantially increased the numberof infectious viral particles produced relative to the HCV RNAlevels.
To characterize secreted viral particles further, we examined thebuoyant density of extracellular infectious Con1/JFH1 wild-type, NS2(L839S) mutant viral particles and All 6 viral particles on an iodixanalequilibrium density gradient. Gradient fractions were collected aftercentrifugation and analyzed for infectivity (Fig. 7). The wild-typevirus exhibited very little infectivity throughout the gradient, as
expected. The NS2 mutant (L839S) and All 6 viruses both displayedinfectious particles ranging in density from 1.05 to 1.19 g/ml (Fig. 7).However, the NS2 mutant exhibited a narrow infectivity peak at a den-sity of 1.15 g/ml, while the All-6 virus showed a broad infectivity peakbetween 1.05 and 1.14 g/ml (Fig. 7).
The adapted Con1/JFH1 virus can be used to evaluate HCV inhibitors
To evaluate the adapted Con1/JFH1 infectious system for antiviralinhibitor screening, we assessed the response of this virus to knownHCV inhibitors. 2′C-methyl adenosine (2′CMA), a nucleoside replica-tion inhibitor, α-CD81 monoclonal antibody, a host-targeted entryinhibitor, and EI-1 (Baldick et al., 2010), a genotype 1-specificentry inhibitor, were tested in a 96-well antiviral assay using theadapted Con1/JFH1 virus and an adapted infectious J6/JFH1 geno-type 2a virus for comparison.

Fig. 7. Characterization of Con1/JFH1 viral particles. Extracellular infectious Con1/JFH1wild-type, NS2 (L839S) mutant and All 6 viral particles were fractionated by equilibriumcentrifugation on a 10–40% iodixanol gradient. Fractions were collected starting fromthe top of the gradient and analyzed by NS3–4A protease activity for infectivity. The pro-files of infectivity are shown for Con1/JFH1wild-type (●); NS2 (L839S) ( ); and All 6 (■).All data were generated from four separate experiments and expressed as mean±SEM.
230 K. Chan et al. / Virology 422 (2012) 224–234
497-5 Lunet cells were infected with either adapted Con1/JFH1 orJ6/JFH1 viruses at an MOI of 0.3. Compounds were added immediatelyafter and incubated for 72 h, after which NS3–4A protease activitywas measured to determine virus replication levels. 50% effectiveconcentrations (EC50 values) are shown in Table 1. The EC50 valuesfor replication inhibitor 2′CMA were comparable for both viruseswhich was expected as both chimeras comprised JFH1-derived repli-case proteins. As an entry inhibitor, α-CD81 antibody efficiently neu-tralized viral infectivity of Con1/JFH1 with an EC50 comparable to thatof the genotype 2a virus. EI-1 was a potent inhibitor of Con1/JFH1 in-fection, with an EC50 of 0.004±0.002 μM. Consistent with previouslypublished results (Baldick et al., 2010), EI-1 had no activity againstthe 2a virus (the EC50 value was >41±15 μM). These results confirmthat the infectious Con1/JFH1 virus system is sensitive to replicationinhibitors and capable of identifying inhibitors that target genotype1b structural proteins.
Discussion
In this study, an intergenotypic 1b/2a hepatitis C virus was suc-cessfully adapted in a novel Huh-7 Lunet subclone. The adaptedviral genome achieved viral titers 1000-fold higher than the parentalstrain. One of the key factors in achieving this adaptation was the useof the highly HCV-permissive 497-5 Lunet cell clone. Although Huh-7Lunet cells support efficient replication and virus production, theirlimited support of virus infection/spread makes it difficult to adaptviruses. The 497-5 Lunet subclone maintains the transfection andreplication efficiencies of Huh-7 Lunet cells but is substantially morepermissive to HCV infection; together, these properties facilitated thepropagation of the 1b/2a chimera in transfected cells until variantsthat supported higher levels of virus production emerged.
Six potential adaptive mutations were identified in the adaptedvirus. One mutation resided in the structural region (core); theother five mutations resided in the non structural region (NS2, NS3protease, NS3 helicase, NS5A and NS5B). Introduction of these six
Table 1Evaluation of the antiviral activity of HCV inhibitors using the adapted Con1/JFH1 virus.
Compound Inhibitor class EC50 and virus genotype Cytotoxicity
Con1/JFH1 J6/JFH1 CC50
Anti-CD81(μg/ml)
Host receptorbinding antagonist
0.09±0.03 0.03±0.01 >2.5
2′CMA (μM) Replication inhibitor 0.35±0.16 0.35±0.19 >50EI-1 (μM) 1b specific entry inhibitor 0.004±0.002 >41±15 >50
mutations into the parental backbone confirmed that these mutationswere responsible for the adapted phenotype. Further investigationthrough addition or removal of individual mutations revealed thatthe NS2 mutation was by far the most critical for improving produc-tion of infectious virus. However, the NS2mutation alone was not suf-ficient to confer the full adapted phenotype as titers of the NS2mutant virus were generally about 10-fold lower than the virus con-taining all 6 mutations. We generated multiple combination mutants,including NS2 mutants in combination with the NS3 helicase, NS5Aor NS5B mutations; these combination mutants achieved high infec-tivity titers but could not consistently produce the peak titers of theAll 6 clone in transfected cells (data not shown). Therefore, we con-clude that the other five mutations (core, NS3, NS5A and NS5B) pro-duce minor but incremental improvements in viral titer on top of theNS2 mutation.
The NS2 L839S mutation may adapt 1b/2a HCV by enhancing infectiousparticle stability
To investigate the mechanism(s) by which the NS2 mutation en-hanced the release of infectious virus particles, we first examinedRNA replication. Intracellular RNA replication was not changed bythe presence of the NS2 mutation, either alone, or in any combinationwith the other five mutations. Furthermore, the amount of RNA se-creted by infected cells did not differ between viral constructs. Thus,the adaptive mutations did not improve infectious virus productionby altering the efficiency of genome replication or enhancing theamount of total RNA genomes secreted by infected cells. In contrast,the specific infectivity of the extracellular and intracellular HCV parti-cles was significantly increased by the NS2 mutation compared to theparental clone or the NS2 revertant clone.
In addition, infectious particles were observed at a range ofrelatively low densities with the NS2 mutation and even more mark-edly when all 6 mutations were present (Fig. 7). Typically, lowerdensity particles are more infectious than higher density particles(Lindenbach et al., 2005). It has been proposed that viral particles inthe low density fractions are associated with lipids and lipoproteins,which are essential elements in the production of infectious HCV par-ticles (Joyce and Tyrrell, 2010; Popescu and Dubuisson, 2010). Basedon the totality of these data, it appears that the unadapted Con1/JFH1chimera was impaired in the assembly of infectious particles. The in-troduction of the adapted NS2 mutation alone substantially restoredthis step, as the assembly or accumulation of infectious particleswas greatly enhanced. The total number of viral genomes synthesizedand secreted was not altered; rather a greater fraction of viral ge-nomes were infectious when the NS2 mutation was present.
Because the NS2 L839S mutation leads to a greater number of in-fectious viral particles, we determined if this mutation could enhanceinfectious particle stability. We observed that the adapted Con1/JFH1virus had an infectious half-life of approximately 5 h (data notshown). This is similar to a J6/JFH1 clone that was recently adaptedin our lab but much longer than an unadapted J6/JFH1 clone (half-life of 1.8 h) (Pokrovskii et al., 2011). Therefore, it is possible thatNS2 L839S results in the formation of longer-lived infectious virions.This would lead to an accumulation of infectious virus and higheroverall titers (Pokrovskii et al., 2011). Unfortunately, we were notable to compare the half-life of the adapted Con1/JFH1 virus to thatof the unadapted wild-type Con1/JFH1, since the titers of the wild-type virus were too low.
The NS2 L839S mutation may alter HCV assembly to increase infec-tious stability. It has been previously shown that NS2 K840A, located inthe loop between transmembrane domains 1 and 2, disrupts a physicalinteraction betweenNS2 andNS3 (Stapleford and Lindenbach, 2010). Inaddition, the NS2 K840A mutation prevents the assembly of infectiousHCV particles (Phan et al., 2009). Interestingly, a mutation in the NS3helicase (Q1247L) partially restores the NS2–NS3 physical interaction

Fig. 8. Location of critical NS2 mutation (L839S). A schematic model of the putative transmembrane domain of NS2 is shown using the one-letter amino acid code. Shaded circlesdenote the NS2 sequence derived from genotype 1b (Con1) while clear circles represent genotype 2a (JFH1) residues. Amino- and carboxy-terminal ends of NS2 are labeled H2Nand CO2H, respectively. The position of Leu 839 is highlighted in yellow and the key adaptive mutation at that position is labeled accordingly. The NS2 catalytic domain is shown as aribbon diagram.
231K. Chan et al. / Virology 422 (2012) 224–234
(Stapleford and Lindenbach, 2010) and fully suppresses theHCV assem-bly defect (Phan et al., 2009). In this work, the NS2 L839Smutation pro-motes the production of long-lived infectious particles, relative to aJFH1 wild-type virus (Pokrovskii et al., 2011). NS2 L839S is also locatedin the linker region between transmembrane domains 1 and 2 (Fig. 8).Therefore, the NS2 L839S mutation may enhance or otherwise alter theNS2–NS3 physical interaction leading to the assembly of longer-livedinfectious particles. Notably, one of the six adaptivemutations observedin this work was an NS3 helicase mutation (I1312V). However, the fulladaptation of the 1b/2a virus requiredmore than just the addition of theNS2 L839S and NS3 helicase I1312V (data not shown). This suggeststhat other mechanisms were required to fully adapt the 1b/2a chimericvirus.
NS2 mutations are required to adapt other chimeric hepatitis C viruses
Similar mutations in NS2 have been seen in previous adapted vi-ruses suggesting that the mechanisms underlying this cell-culture ad-aptation are conserved across genotypes. In previous studies for 1a —
(H77S strain); 4a — (ED43 strain) and 1b — (J4 strain) JFH1 chimericviruses, production of infectious virus also depended on a compen-satory mutation in NS2, especially for chimeric viruses where the junc-tion is located within NS2 (Gottwein et al., 2009; Scheel et al., 2008; Yiet al., 2007). In a study of 1a (H77S)/JFH1 chimeric viruses, differentamino acid substitutions were observed at the same residue in the 1aportion of NS2 (I839T). However, the insertion of this mutation(I839T) into a H77S/JFH1 chimera, where the junction was not locatedinternally in NS2 but at the NS2/NS3 boundary, abolished release ofinfectious virus by this alternative chimera. Similar findings were ob-served in a study of genotype 4a (ED43)/JFH1 chimeric virus (Scheelet al., 2008), except this mutation was located slightly upstream at res-idue 827 of NS2 (T827A). This mutation greatly enhanced virus produc-tion but only when the intergenotypic junctionwas locatedwithin NS2.An additional mutation in NS2 at residue 977 (T977S) was requiredto enhance virus production in a 4a/JFH1 chimeric virus with thejunction located at the NS2/NS3 boundary. In additional studies notdescribed here, the L839S mutation in NS2 also abolished virus produc-tion in a Con1/JFH1-All 6 chimera with an intergenotypic junctionlocated in the NS2/NS3 boundary. Interestingly, removal of the L839Smutation in this chimera rescued virus production (data not shown).
Although an adapted mutation in NS2 (G838R) emerged in vivofrom chimpanzees inoculated with JFH1 (Kato et al., 2008), mutationsin NS2 were not prevalent or critical in JFH1 cell culture adaptationstudies (Delgrange et al., 2007; Kaul et al., 2007). It has been suggestedthat these mutations in NS2 are chimera specific and may correct forincompatibilities between proteins of these different HCV genotypesat sites essential for protein–protein interactions (Yi et al., 2007). There-fore the L839Smutation in the transmembrane loop of NS2may correctan incompatibility that exists between Con1 and JFH1 proteins andimprove assembly of infectious particles.
The HCV 1b/2a as a tool for developing HCV inhibitors
Development of full-length HCV genomes is a valuable tool foridentification of inhibitors targeting all steps of the viral life cycle. Al-though, the discovery of HCV replicons was a major achievement,they only permit studies of RNA replication. A virus-based systemwill facilitate the identification of molecules targeting additional pro-cesses including viral entry, assembly and egress. Using the genotype1 specific E2 inhibitor, EI-1 (Baldick et al., 2010), we confirmed thatthe adapted Con1/JFH1 clone could identify genotype 1b active com-pounds that were inactive against genotype 2 viruses. Overall, we an-ticipate that, based on the high titer that it achieves, the adapted1b/2a chimeric virus reported here will facilitate both drug discoveryand molecular virology efforts.
Materials and methods
Cell culture
Huh-7 Lunet cells were obtained from ReBLikon GmbH (Mainz,Germany) (Blight et al., 2002). Lunet-CD81 cells were generated bytransducing Huh-7 Lunet cells with a lentivirus expressing human-CD81 followed by selection using DMEM complete medium (seebelow) supplemented with blasticidin (5 μg/ml) (Pokrovskii et al.,2011). 497-5 Lunet cells were derived from Huh-7 Lunet cells andwere obtained from patient-derived NS5B/1b-Con1 chimeric repliconin vitro resistance selection studies. All cell lines described weremaintained in Dulbecco's modified Eagle's medium (DMEM) withGlutaMAX-I (Invitrogen, Carlsbad, CA) supplemented with 10% fetal

232 K. Chan et al. / Virology 422 (2012) 224–234
bovine serum (Hyclone, Logan, UT), 100 U/ml of penicillin (Invitrogen),100 μg/ml of streptomycin (Invitrogen) and 10 mM non-essentialamino acids (Invitrogen). All cell lines were maintained in humidifiedincubators at 37 °C and 5% CO2.
Plasmid construction
pCon1/JFH1The plasmid Con1/JFH1 virus was constructed from plasmids pCon1
(Apath LLC., Brooklyn, NY) and pJFH1(Toray Inc., Japan) using genesplicing by overlap extension PCR as described by Zhang et al. (2008)with slight modifications. PCR was performed using High Fidelity PCRMaster (Roche Diagnostics Corporation, Indianapolis, IN) following themanufacturer's suggested protocol. Four PCRswere performed to gener-ate an EcoRI–SpeI fusion product which contained: JFH1 5′untranslatedregion (UTR); Con1 core, E1, E2, p7 and the first 33 amino acids of NS2,followed by the rest of NS2 from JFH1 up to NS3. The first PCR, primers2aEcoRI_fw (5′ GAC GTT GTA AAA CGA CGG CCA GTG GAA TTC 3′) and2a-1bcore_rev (5′GAA CTT GAC GTC CTG TGG GCG ACG GTT GGT GTTTCT TTTGG 3′) was used to amplify EcoRI, 5′UTR plus thefirst 19 codonsof core from pJFH1. The second PCR, primers 2a-1bcore_fw (5′ CCA AAAGAA ACA CCA ACC GTC GCC CAC AGG ACG TCA AGT TC 3′) and 1bNS2(C3)-2a_rev (5′AGA TAG CAC AAC CAC CAC AGG AGC CTA GCG AGGAAC AGC TTA T 3′) was used to amplify pCon1b in the region of coreup to the first 33 amino acids of NS2. The third PCR, primers 1bNS2(C3)-2a_fw (5′ GCT GTT CCT CGC TAG GCT CCT GTG GTG GTT GTG CTATCT CC 3′) and 2a(4541–4570)_rev (5′CCC AAA CCC CAG GGT GGCAGC TAC CGA GGG 3′) was used to amplify pJFH1 in the region ofNS2 starting at its 34th amino acid up to the SpeI site in NS3. In thefourth and final PCR, the previous three PCR products were combinedusing primers 2aEcoRI_fw and 2a(4541–4570)_rev to generate thefinal EcoRI–SpeI PCR product. The final product was digested withEcoRI andSpeI and introduced into pJFH1 to generate theCon1/JFH1chi-meric HCV genome. For construction of adapted Con1/JFH genomes,amino acid substitutions were introduced by site-directed mutagenesisusing a Quick Change Site Directed Mutagenesis kit (Agilent Technolo-gies, Santa Clara, CA). All final constructs were verified by DNA sequenc-ing (Elim Biopharmaceuticals Inc, Hayward, CA).
pJc-1 RlucFirst pJc-1 was constructed from plasmids pJ6/JFH1 (Pokrovskii et
al., 2011) and pJFH1 using gene splicing by overlap extension PCR as de-scribed above. Three PCRs were performed to generate an EcoRI–AflIIfusion product which contained, JFH1 5′untranslated region (UTR);J6 core, E1, E2, p7 and the first 33 amino acids of NS2, followed by therest of NS2 from JFH1 up to the AflII site. The first PCR, primers pUC19E-coRIup_fw (5′ GCT GCA AGG CGA TTA AGT TGG GTA ACG 3′) andJ6NS2C3JFH1_rev (5′ GAG ATA GCA CAA CCA CCA CAG ACA CTG AAACCG GCT GAG AAG GGT CTT ATA CCC 3′) was used to amplify EcoRIup to the first 33 amino acids of NS2 of J6 from pJ6/JFH1. The secondPCR, primers C3JFH1-NS2_fw (5′ CAG TGT CTG TGG TGG TTG TGC TATCTC CTG) and JFH1-NS2AflIIdown_rev (5′ CCA AAG CGC ATA CCC TTATCA GAG CGT GAG 3′) was used to amplify NS2 of JFH1 starting at its34th amino acid up to the AflII site in NS2. In the final PCR the previoustwo PCR productswere combined using primers pUC19EcoRIup_fw andJFH1-NS2AflIIdown_rev to generate the final EcoRI–AflII PCR product.The final product was digested with EcoRI and AflII and introducedinto pJFH1 to generate the pJc-1 HCV genome. The reporter hRlucgene fused to the 2A protease of foot and mouth disease virus (FMDV)was inserted into pJc-1 as described by Zhang et al. (2008) to constructpJc-1 Rluc. Four PCRs were performed to generate an EcoRI–BglII fusionproduct which contained the JFH1 5′untranslated region (UTR); hRluc,FMDV, followed by J6 core to E2. The first PCR, primers 3′FMDV 2aPacI_J6 5′core_fw (5′ CGA GTC CAA CCC TGG GCC CTT AAT TAA CAGCAC AAA TCC TAA ACC 3′, restriction site PacI underlined) and J6E2-BglII_rev (5′ AGT CGA CAA GGC GGG CAG ATC TGA GTA AGA GCA
AGG TAA AAT GG 3′) was used to amplify pJc-1 in the region of coreto E2 up to the BglII site. The second PCR, primers J6Core17-PmeI-hRLuc_fw (5′ CCA AAA GAA ACA CCA ACC GTT TAA ACG CTT CCA AGGTGT ACG ACC CCG 3′, restriction site PmeI underlined) and 3′hRLucFMDV2a_rev (5′ GGT CAA AAT TCA ACA GCT GCT GCT CGT TCTTC AGC 3′) was used to amplify the hRluc gene from pF9 cytomegalo-virus (CMV) hRluc-Neo Flexi(R) (Promega, Madison, WI). In the samePCR, the primers FMDV2a_fw (5′ CAG CTG TTG AAT TTT GAC CTT CTTAAG CTT GCG GGA GAC GTC GAG TCC AAC CCT GGG CCC 3′) and 3′FMDV2a-PacI-J6 5′core_rev (5′ GGT TTA GGA TTT GTG CTG TTA ATTAAG GGC CCA GGG TTG GAC TCG 3′) which contain the sequence ofFMDV were included to generate a fusion between the hRluc gene andthe sequence of FMDV. The third PCR, primers EcoRI-2a 5′UTR_fw (5′ACC GAA ACG CGC GAG AAT TCT AAT ACG ACT CAC TAT AGA CC 3′, re-striction site EcoRI underlined) and J6Core17-PmeI-hRLuc_rev (5′ CGGGG TCG TAC ACC TTG GAA GCG TTT AAA CGG TTG GTG TTT CTTTTG G 3′, restriction site PmeI underlined) was used to amplify pJc-1in the region of EcoRI 5′UTR plus the first 17 codons of core. In thefinal PCR, the previous three PCR products were combined usingprimers EcoRI-2a 5′UTR_fw and J6E2-BglII_rev to generate the finalEcoRI- BglII PCR product. The final product was introduced into pJc-1to generate pJc-1 Rluc after digestion with EcoRI and BglII.
pJ6/JFH1The plasmid pJ6/JFH1 was previously described (Pokrovskii et al.,
2011).
RNA transcription
Plasmids were linearized with Xba I and purified using a PCR purifi-cation kit (Qiagen, Valencia, CA). RNA was synthesized with T7 MEGA-Script reagents (Ambion, Austin, TX) following the manufacturer'ssuggested protocol and reactions were stopped by digesting withRNase free DNase. RNA was purified using an RNA easy kit (Qiagen) inaccordance with the manufacturer's protocol. The quality of synthe-sized RNA was confirmed by agarose gel electrophoresis and RNAconcentrationswere determined bymeasurement of the optical densityat 260 nm.
Electroporation of HCV RNAs
497-5 Lunet cells were trypsinized and washed twice withphosphate-buffered saline (PBS)without Ca2+ orMg2+ and then resus-pended at 107 cells/ml in reduced serum Opti-MEM (Invitrogen). 5 or10 μg of in vitro RNA transcripts was mixed with 0.4 ml cell suspensionin a Gene Pulser cuvette (0.4 cm gap) and electroporated at 270 V and960 μF using a Gene Pulser system (Bio-Rad, Hercules, CA). Immediatelyafter electroporation, cells were resuspended in 25 ml complete DMEMmedium and seeded as required for assays.
Adaptation of Con1/JFH1 to 497-5 Lunet cells
497-5 Lunet cells were electroporated as described above with5 μg Con1/JFH1 wild-type RNA and seeded into 75 cm2
flasks. After96 h post transfection, cells were transferred into larger 162 cm2
flasks. Cells were passaged at a dilution of 1:3 every 2 to 3 days andmonitored for presence of HCV in cells by NS5A immunostainingand in supernatants by TCID50 assay at the indicated time points.When the cells were >80% infected with HCV, virus containing super-natants was used to infect naïve 497-5 Lunet cells followed by furthercell passaging until sufficient virus spread and infectivity wasachieved by the adapted virus.

233K. Chan et al. / Virology 422 (2012) 224–234
Measurement of cell-associated infectivity for HCV-transfected cells
Huh-7 Lunet cells were transfected with HCV RNA transcripts; atthe indicated time points cell culture supernatants were harvested,virus titers were determined by the TCID50 assay and HCV RNA levelswere quantified by quantitative real-time RT-PCR assay. Cell-associated infectivity was prepared as described in Gastaminza et al.(2006). Briefly, cells were washed with PBS, trypsinized and resus-pended in cell culture media. Cells were lysed by four freeze thawcycles in dry ice and 37 °C water bath. Cell debris was pelleted bycentrifugation for 3 min at high speed. Supernatant was collectedand used for infection of naïve cells and HCV RNA quantification.
Indirect immunofluorescence staining for HCV NS5A protein
Infected cells seeded in a 96 well black plate were fixed with4% paraformaldehyde at room temperature for 10 min. Cells werethen washed with PBS and incubated with blocking solution con-taining 3% BSA, 0.3% Triton X-100, and 10% FBS for at least 30 min.Cells were then incubated with a mouse monoclonal antibody toHCV NS5A protein (9E10, Apath, Brooklyn, NY) at a dilution of1:10,000 in PBS with 3% BSA, 0.3% Triton X-100 for 1 h at room tem-perature. After three washes with PBS, cells were incubated withsecondary antibody (Alexa-Fluor 555 goat anti-mouse IgG [H+L],Invitrogen) at a dilution of 1:1000 and Hoechst 33342 for DNA nu-clei staining in PBS with 3% BSA, 0.3% Triton X-100 for 1 h. Finally,cells were washed three times in PBS and stained cells were imagedusing a Zeiss microscope with fluorescence capabilities. Percentagesof infected cells were determined using an ImageXpress Micro(Molecular Devices, Sunnyvale, CA) with MetaExpress 2.0 softwareby comparing cells stained positive with anti-NS5A antibody versusHoechst stained nuclei.
Determination of virus titers in cell culture supernatants
497-5 Lunet cells were seeded in black clear bottom 96-well platesat a density of 5000 cells per well in 100 μl DMEM culture medium.After overnight incubation cells were infected with one- to four-foldserial dilutions of virus containing cell culture supernatant, withfour replicates per dilution. After 72 h cells were stained for NS5A asdescribed. Virus titer was determined by calculating the 50% tissueculture infectious dose (TCID50/ml) according to the method of Reedand Muench (1938). Wells were scored positive if at least one NS5Apositive cell was detected.
Quantification of HCV RNA by real time RT-PCR
HCV RNA from cell lysates or cell culture supernatants was iso-lated as described above. Quantitative real time RT-PCR analysis wascarried out using a one step RT-PCR kit, QuantiTect Multiplex RT-PCR kit (Qiagen). Quantitative RT-PCR was carried out for HCV andglyceraldehyde-3-phosphate dehydrogenase (GAPDH), an internalcontrol, according to the manufacturer's instructions. Briefly, 4 μl ofRNA was reverse-transcribed in a 25 μl reaction mix at 44 °C for30 min, followed by inactivation of reverse-transcriptase at 95 °C for10 min. Products were amplified by PCR in 38 cycles each that com-prised 95 °C for 15 s and 58 °C for 30 s. HCV primers targeting JFH1NS3 were: HCV forward primer, 5′-CACGGAGCTGGCAACAAGACT-3′, HCV reverse primer, 5′-G-FAM-CAGTGCGTCTACATGAGCCTGGC-3′.GAPDH primers were purchased from Invitrogen, Human GAPDH —
Certified LUX™ Primer Set (Invitrogen) labeled with FAM. HCV andGAPDH RNA quantifications were based on a standard curve, the signalof HCV RNA was normalized to that of GAPDH RNA at each time point.
Extraction and isolation of HCV RNA for genotypic analysis
HCV RNA from cells infected with virus or cell culture supernatantwas isolated using RNeasy and QiaShredder kits or QiaAmp Viral RNAkit (all kits from Qiagen) according to the manufacturer's protocols.First strand cDNA synthesis from isolated RNA was performed with theSuperScript®III First-Strand Synthesis Supermix System (Invitrogen).Seven overlapping fragments of the first strand cDNA were amplifiedby PCR using the High Fidelity PCRMaster kit (Roche Diagnostics Corpo-ration, Indianapolis, IN). DNA sequencing was performed by ElimBiopharmaceuticals Inc.
Luciferase assay
Huh-7 Lunet, Lunet-CD81 or 497-5 Lunet cells were seeded in 96well plates at a density of 5000 cells perwell. After overnight incubationcells were infected with 100 μl of viral inocula (Jc-1 Rluc). Following3 days of incubation, cell culture medium was removed and cells werelysed and assayed for luciferase activity as a marker for intracellularHCV replication levels. Luciferase activity was measured with Renillaluciferase assay buffer and substrate (Promega), following themanufac-turer's protocols, using a VICTOR3™VMultilabel Counter (Perkin Elmer,Shelton, CT).
Intracellular NS3–4A protease activity
NS3–4A protease activity was used to monitor intracellular HCVreplication levels and was measured using a europium labeled NS3–4A protease substrate as described by Yang and Delaney (2006),with slight modifications. In brief, media were removed from virusinfected cells and replaced with 50 μl of a lysis-NS3 substrate solutioncontaining: 1× Promega lysis buffer; 150 mM NaCl, and 150 nM NS3europium substrate (AnaSpec, Freemont, CA) in water. Time resolvedfluorescence was measured using a VICTOR3™ V Multilabel Counter(Perkin Elmer).
Iodixanol density gradient analysis of cell culture supernatants
To determine the buoyant densities of viral particles aliquots ofextracellular medium were separated using equilibrium density gra-dient ultracentrifugation according to the method of Lindenbach etal. (2005). Briefly, 1 ml of extracellular medium was loaded at thetop of a 10 ml 10–40% iodixanol gradient. Iodixanol gradients wereprepared by using 10% and 40% iodixanol solutions that each con-tained 10 mM HEPES, pH 7.5, and 0.02% BSA. In addition, the 10%iodixanol solution contained 125 mM NaCl and the 40% iodixanolsolution contained 50 mM NaCl to maintain iso-osmolarity. Sampleswere centrifuged through the gradients at 40,000 rpm for 16 h at4 °C in an SW41 Ti swinging bucket rotor (Beckman, Brea, CA).0.4 ml fractions from each gradient were collected from the top.Each fraction was assayed for relative infectivity using the NS3–4Aprotease assay described above.
Antiviral and neutralization assays
497-5 Lunet cells were seeded in white clear bottom 96 wellplates at a density of 5000 cells per well. After overnight incubation,cells were infected with 100 μl of viral inocula at an MOI of 0.3.Three-fold serial drug dilutions were prepared in DMSO and addedto cells at a 1:200 dilution achieving a final concentration of 0.5%DMSO in a final volume of 200 μl. The final concentrations of drugranged from 2.5 to 50,000 nM for both 2′CMA (Acme Bioscience,Belmont, CA) and E-I (Chembridge, San Diego, CA). Anti-CD81-monoclonal antibody JS-81 (BD Biosciences, San Jose, CA) was seriallydiluted in DMEM yielding final concentrations from 1 ng/ml to2500 ng/ml. Following 3 days of incubation, NS3 protease activity

234 K. Chan et al. / Virology 422 (2012) 224–234
was used to quantify intracellular HCV replication levels as describedabove.
Modeling membrane-associated NS2
A schematic model of the transmembrane domain of NS2 was con-structed (Fig. 8) using the program LaTeX (www.latex-project.org)and the associated macro package, TEXtopo (Beitz, 2000). Transmem-brane residues were selected manually and the selection criteria werebased in part on the NS2 membrane topology analysis of Yamaga andOu (2002). A three-dimensional model of the catalytic domain wasmanually placed at the C-terminal region of the predicted transmem-brane domain and was derived from the coordinates of the HCV geno-type 1a NS2 crystal structure (PDB ID: 2HD0) (Lorenz et al., 2006).
Acknowledgments
The authors gratefully acknowledge Zach Newby for helpful dis-cussions and advice on molecular modeling. We also thank MatthewPaulson for helpful discussions and Linda Slanec Higgins for criticalreview of this manuscript.
References
Baldick, C.J., et al., 2010. A novel small molecule inhibitor of hepatitis C virus entry.PLoS Pathog. 6.
Bartenschlager, R., Pietschmann, T., 2005. Efficient hepatitis C virus cell culture system:what a difference the host cell makes. Proc. Natl. Acad. Sci. U. S. A. 102, 9739–9740.
Beitz, E., 2000. T(E)Xtopo: shaded membrane protein topology plots in LAT(E)X2epsilon.Bioinformatics 16, 1050–1051.
Blight, K.J., et al., 2002. Highly permissive cell lines for subgenomic and genomic hep-atitis C virus RNA replication. J. Virol. 76, 13001–13014.
Delgrange, D., et al., 2007. Robust production of infectious viral particles inHuh-7 cells by in-troducingmutations in hepatitis C virus structural proteins. J. Gen. Virol. 88, 2495–2503.
Gastaminza, P., et al., 2006. Differential biophysical properties of infectious intracellu-lar and secreted hepatitis C virus particles. J. Virol. 80, 11074–11081.
Gottwein, J.M., et al., 2007. Robust hepatitis C genotype 3a cell culture releasing adaptedintergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology 133, 1614–1626.
Gottwein, J.M., et al., 2009. Development and characterization of hepatitis C virus geno-type 1–7 cell culture systems: role of CD81 and scavenger receptor class B type Iand effect of antiviral drugs. Hepatology (Baltimore, Md.) 49, 364–377.
Joyce, M.A., Tyrrell, D.L.J., 2010. The cell biology of hepatitis C virus. Microbes Infect. 12,263–271.
Kato, T., et al., 2008. Hepatitis C virus JFH-1 strain infection in chimpanzees is associatedwith low pathogenicity and emergence of an adaptive mutation. Hepatology(Baltimore, Md.) 48, 732–740.
Kaul, A., et al., 2007. Cell culture adaptation of hepatitis C virus and in vivo viability ofan adapted variant. J. Virol. 81, 13168–13179.
Koutsoudakis, G., et al., 2007. The level of CD81 cell surface expression is a key deter-minant for productive entry of hepatitis C virus into host cells. J. Virol. 81, 588–598.
Lanford, R.E., et al., 2009. The accelerating pace of HCV research: a summary of the 15thInternational Symposium on Hepatitis C Virus and Related Viruses. Gastroenterology136, 9–16.
Lindenbach, B.D., et al., 2005. Complete replication of hepatitis C virus in cell culture.Science 309, 623–626.
Lorenz, I.C., et al., 2006. Structure of the catalytic domain of the hepatitis C virus NS2–3protease. Nature 442, 831–835.
Mangia, A., Andriulli, A., 2008. Are HCV genotypes 2 and 3 the same or different? Curr.Hepat. Rep. 7, 88–92.
Miyamoto, M., et al., 2006. Comparison between subgenomic replicons of hepatitis Cvirus genotypes 2a (JFH-1) and 1b (Con1 NK5.1). Intervirology 49, 37–43.
Pawlotsky, J.M., et al., 2007. The hepatitis C virus life cycle as a target for new antiviraltherapies. Gastroenterology 132, 1979–1998.
Phan, T., et al., 2009. Hepatitis C virus NS2 protein contributes to virus particle assemblyvia opposing epistatic interactions with the E1–E2 glycoprotein and NS3–NS4Aenzyme complexes. J. Virol. 83, 8379–8395.
Pietschmann, T., et al., 2006. Construction and characterization of infectious intrageno-typic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A.103, 7408–7413.
Pokrovskii, M.V., et al., 2011. Novel mutations in a tissue culture-adapted hepatitis Cvirus strain improve infectious-virus stability and markedly enhance infectionkinetics. J. Virol. 85, 3978–3985.
Popescu, C.I., Dubuisson, J., 2010. Role of lipid metabolism in hepatitis C virus assemblyand entry. Biol. Cell. 102, 63–74.
Reed, L.J., Muench, H., 1938. A simple method of estimating 50 per cent end-points. Am.J. Hyg. 27, 493–497.
Scheel, T.K., et al., 2008. Development of JFH1-based cell culture systems for hepatitis Cvirus genotype 4a and evidence for cross-genotype neutralization. Proc. Natl. Acad.Sci. U. S. A. 105, 997–1002.
Stapleford, K.A., Lindenbach, B.D., 2010. Hepatitis C virus NS2 coordinates virus particleassembly through physical interactions with the E1–E2 glycoprotein and NS3–NS4A enzyme complexes. J. Virol. 85, 1706–1717.
Wakita, T., et al., 2005. Production of infectious hepatitis C virus in tissue culture from acloned viral genome. Nat. Med. 11, 791–796.
WHO, 1999. Global surveillance and control of hepatitis C. Report of a WHO Consulta-tion organized in collaboration with the Viral Hepatitis Prevention Board, Antwerp,Belgium. J. Viral Hepat. 6, 35–47.
Woerz, I., et al., 2009. Hepatitis C virus replicons: dinosaurs still in business? J. ViralHepat. 16, 1–9.
Yamaga, A.K., Ou, J.H., 2002. Membrane topology of the hepatitis C virus NS2 protein.J. Biol. Chem. 277, 33228–33234.
Yang, H., Delaney IV, W.E., 2006. A novel fluorescence-based protease assay using theendogenous NS3/4A protease activity present in the total cell lysates of HCV repliconcells. J. Clin. Virol. 36, S109.
Yi, M., et al., 2007. Compensatory mutations in E1, p7, NS2, and NS3 enhance yieldsof cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 81,629–638.
Zhang, Y., et al., 2008. Novel chimeric genotype 1b/2a hepatitis C virus suitable forhigh-throughput screening. Antimicrob. Agents Chemother. 52, 666–674.