aerobic exercise training–induced left ventricular...
TRANSCRIPT

Aerobic Exercise Training–Induced Left VentricularHypertrophy Involves Regulatory MicroRNAs, Decreased
Angiotensin-Converting Enzyme-Angiotensin II, andSynergistic Regulation of Angiotensin-Converting Enzyme
2-Angiotensin (1-7)Tiago Fernandes, Nara Y. Hashimoto, Flavio C. Magalhaes, Fernanda B. Fernandes, Dulce E. Casarini,
Adriana K. Carmona, Jose E. Krieger, M. Ian Phillips, Edilamar M. Oliveira
Abstract—Aerobic exercise training leads to a physiological, nonpathological left ventricular hypertrophy; however, theunderlying biochemical and molecular mechanisms of physiological left ventricular hypertrophy are unknown. The role ofmicroRNAs regulating the classic and the novel cardiac renin-angiotensin (Ang) system was studied in trained rats assignedto 3 groups: (1) sedentary; (2) swimming trained with protocol 1 (T1, moderate-volume training); and (3) protocol 2 (T2,high-volume training). Cardiac Ang I levels, Ang-converting enzyme (ACE) activity, and protein expression, as well as AngII levels, were lower in T1 and T2; however, Ang II type 1 receptor mRNA levels (69% in T1 and 99% in T2) and proteinexpression (240% in T1 and 300% in T2) increased after training. Ang II type 2 receptor mRNA levels (220%) and proteinexpression (332%) were shown to be increased in T2. In addition, T1 and T2 were shown to increase ACE2 activity andprotein expression and Ang (1-7) levels in the heart. Exercise increased microRNA-27a and 27b, targeting ACE anddecreasing microRNA-143 targeting ACE2 in the heart. Left ventricular hypertrophy induced by aerobic training involvesmicroRNA regulation and an increase in cardiac Ang II type 1 receptor without the participation of Ang II. Parallel to this,an increase in ACE2, Ang (1-7), and Ang II type 2 receptor in the heart by exercise suggests that this nonclassic cardiacrenin-angiotensin system counteracts the classic cardiac renin-angiotensin system. These findings are consistent with a modelin which exercise may induce left ventricular hypertrophy, at least in part, altering the expression of specific microRNAstargeting renin-angiotensin system genes. Together these effects might provide the additional aerobic capacity required by theexercised heart. (Hypertension. 2011;58:182-189.) ● Online Data Supplement
Key Words: aerobic exercise training � cardiac hypertrophy � renin angiotensin system � microRNAs� angiotensin II receptors � ACE2 � angiotensin (1-7)
Left ventricular hypertrophy (LVH) induced by aerobicexercise training is an important physiological compensa-
tory mechanism in response to chronic increases in hemodynam-ic overload. This phenotype is associated with sarcomeres addedin series to lengthen the cardiac cell, as well as in parallel. Theincreased cross-sectional area contributes to increased ventricu-lar stroke volume and cardiac output, which improves aerobiccapacity. In contrast, pathological LVH in cardiovascular dis-eases is associated with increased fibrosis and lowered aerobiccapacity, leading to high mortality.1–3
Several studies have reported that the renin-angiotensin(Ang) system (RAS) plays an important role in the progres-sion of LVH.4–6 However, there are only limited data about
the mechanisms of exercise training involved in RAS andLVH. The aim of this study was to elucidate these mecha-nisms of exercise training on physiological LVH.
Pathological LVH occurs with arterial hypertension,7,8
myocardial infarction,9,10 and heart failure.11 These diseasestates are also associated with increased local cardiac RASlevels, represented by augmented angiotensinogen, angioten-sin-converting enzyme (ACE), and angiotensin II (Ang II).Blockade of the classic RAS promotes therapeutic benefits topatients with essential hypertension and cardiovascular dis-ease.8 Cardiac Ang II is implicated in the induction of fibrosisbut is not required for LVH.12 LVH is produced without theparticipation of Ang II in transgenic animal models for RAS
Received December 10, 2010; first decision December 27, 2010; revision accepted May 24, 2011.From the Laboratory of Biochemistry and Molecular Biology of Exercise, School of Physical Education and Sport (T.F., N.Y.H., F.C.M., E.M.O.), and
Laboratory of Genetics and Molecular Cardiology, InCor, Medical School (J.E.K.), University of Sao Paulo, Sao Paulo, Brazil; Nephrology Division,Kidney and Hypertension Hospital (F.B.F., D.E.C.), and Department of Biophysics (A.K.C.), Universidade Federal de Sao Paulo, Sao Paulo, Brazil;Laboratory of Stem Cells (M.I.P., E.M.O.), Keck Graduate Institute, Claremont, CA.
Correspondence to Edilamar M. Oliveira, School of Physical Education and Sport, University of Sao Paulo, Laboratory of Biochemistry and MolecularBiology of Exercise, Av Professor Mello Moraes, 65 Cidade Universitaria, Sao Paulo 05508-900, Brazil. E-mail [email protected]
© 2011 American Heart Association, Inc.
Hypertension is available at http://hyper.ahajournals.org DOI: 10.1161/HYPERTENSIONAHA.110.168252
182
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from
components13–15 and in Ang II type 1 (AT1) receptor activa-tion by mechanical stress.16–18 A novel cardiac RAS includesACE2, which is an essential regulator of heart function19 andplays a pivotal role in Ang (1-7) formation. This novel RASis implicated in vasodilatation and control of fibrosis.20–23
Previous findings suggest that ACE2 maintains the importantbalance between the Ang II and Ang (1-7), favoring cardio-vascular homeostasis. However, the role of exercise trainingin the cardiac ACE2-Ang (1-7) axis is unknown. We haveshown that AT1 receptor blockade prevents physiologicalLVH induced by resistance training18 and by aerobic exercisetraining.24 Moreover, exercise training promoted LVH bycardiac RAS stimulation independent of the systemic RAS.18,24
Several genes are regulated by microRNAs (miRNAs).MiRNAs are endogenous, small, and noncoding RNAs, whichare targeted to specific genes and function as negative regulatorsof gene expression by inhibiting translation or promoting deg-radation of target mRNAs. Recent studies have shown the rolesplayed by miRNAs in different forms of cardiovascular diseaseand pathological LVH.25,26 However, miRNAs may be impor-tant for normal development and in physiological cardiac hyper-trophy induced by aerobic exercise training. In the present study,it was hypothesized that exercise training alters specificmiRNAs that regulate their target cardiac RAS genes and tipthe balance of classic RAS genes in favor of the novel RASgenes to contribute to physiological LVH.
Materials and MethodsAnimal CareAll of the protocols and surgical procedures used were in accordancewith the National Institutes of Health Guide for the Care and Use ofLaboratory Animals and were approved by the ethics committee ofthe University of Sao Paulo School of Physical Education and Sport.Female normotensive Wistar rats (190 to 220 g, n�42) were used.The animals were housed 3 to 5 per cage at a controlled roomtemperature (22°C) with a 12-hour dark-light cycle and fed standardrat chow, having access to water ad libitum.
The rats were randomly divided into 3 experimental groups, eachwith 14 rats: (1) sedentary (S; n�7); (2) swimming trained withprotocol 1 (T1; n�7); and (3) swimming trained with protocol 2 (T2;n�7). Each group was subdivided into 2 groups, one for hemody-namic, biochemical, and molecular studies and the other for mor-phological and histological studies. For the detailed Material andMethods section, please see the online Data Supplement, available athttp://hyper.ahajournals.org.
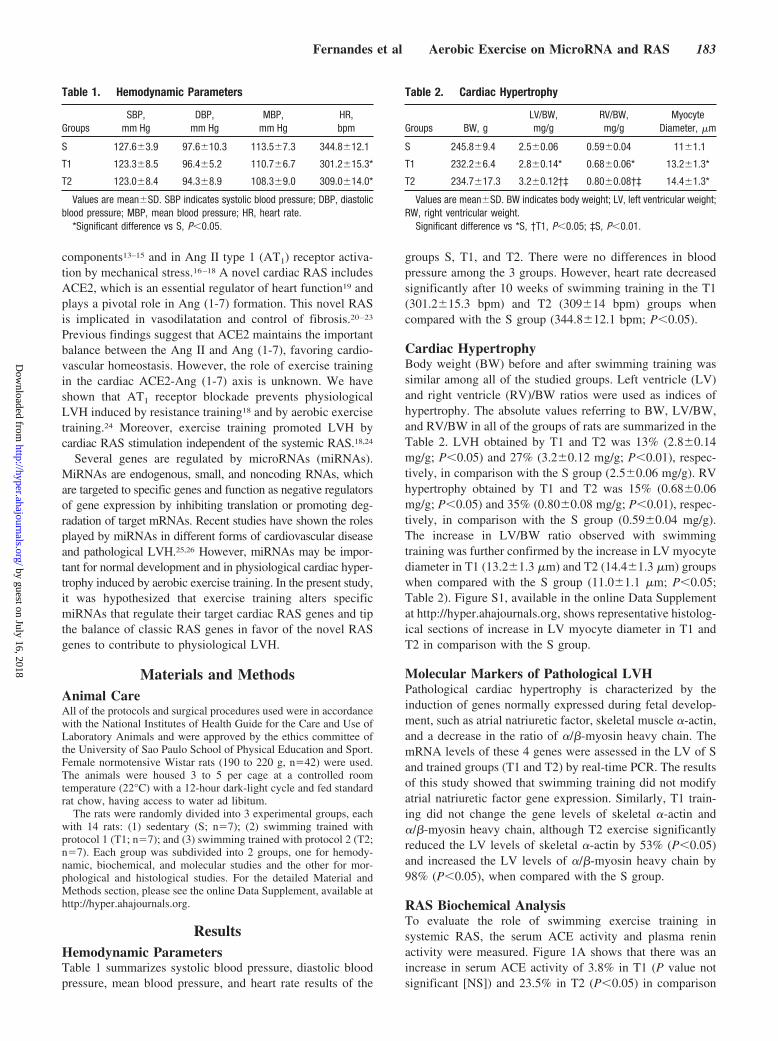
ResultsHemodynamic ParametersTable 1 summarizes systolic blood pressure, diastolic bloodpressure, mean blood pressure, and heart rate results of the
groups S, T1, and T2. There were no differences in bloodpressure among the 3 groups. However, heart rate decreasedsignificantly after 10 weeks of swimming training in the T1(301.2�15.3 bpm) and T2 (309�14 bpm) groups whencompared with the S group (344.8�12.1 bpm; P�0.05).
Cardiac HypertrophyBody weight (BW) before and after swimming training wassimilar among all of the studied groups. Left ventricle (LV)and right ventricle (RV)/BW ratios were used as indices ofhypertrophy. The absolute values referring to BW, LV/BW,and RV/BW in all of the groups of rats are summarized in theTable 2. LVH obtained by T1 and T2 was 13% (2.8�0.14mg/g; P�0.05) and 27% (3.2�0.12 mg/g; P�0.01), respec-tively, in comparison with the S group (2.5�0.06 mg/g). RVhypertrophy obtained by T1 and T2 was 15% (0.68�0.06mg/g; P�0.05) and 35% (0.80�0.08 mg/g; P�0.01), respec-tively, in comparison with the S group (0.59�0.04 mg/g).The increase in LV/BW ratio observed with swimmingtraining was further confirmed by the increase in LV myocytediameter in T1 (13.2�1.3 �m) and T2 (14.4�1.3 �m) groupswhen compared with the S group (11.0�1.1 �m; P�0.05;Table 2). Figure S1, available in the online Data Supplementat http://hyper.ahajournals.org, shows representative histolog-ical sections of increase in LV myocyte diameter in T1 andT2 in comparison with the S group.
Molecular Markers of Pathological LVHPathological cardiac hypertrophy is characterized by theinduction of genes normally expressed during fetal develop-ment, such as atrial natriuretic factor, skeletal muscle �-actin,and a decrease in the ratio of �/�-myosin heavy chain. ThemRNA levels of these 4 genes were assessed in the LV of Sand trained groups (T1 and T2) by real-time PCR. The resultsof this study showed that swimming training did not modifyatrial natriuretic factor gene expression. Similarly, T1 train-ing did not change the gene levels of skeletal �-actin and�/�-myosin heavy chain, although T2 exercise significantlyreduced the LV levels of skeletal �-actin by 53% (P�0.05)and increased the LV levels of �/�-myosin heavy chain by98% (P�0.05), when compared with the S group.
RAS Biochemical AnalysisTo evaluate the role of swimming exercise training insystemic RAS, the serum ACE activity and plasma reninactivity were measured. Figure 1A shows that there was anincrease in serum ACE activity of 3.8% in T1 (P value notsignificant [NS]) and 23.5% in T2 (P�0.05) in comparison
Table 1. Hemodynamic Parameters
GroupsSBP,
mm HgDBP,
mm HgMBP,
mm HgHR,bpm
S 127.6�3.9 97.6�10.3 113.5�7.3 344.8�12.1
T1 123.3�8.5 96.4�5.2 110.7�6.7 301.2�15.3*
T2 123.0�8.4 94.3�8.9 108.3�9.0 309.0�14.0*
Values are mean�SD. SBP indicates systolic blood pressure; DBP, diastolicblood pressure; MBP, mean blood pressure; HR, heart rate.
*Significant difference vs S, P�0.05.
Table 2. Cardiac Hypertrophy
Groups BW, gLV/BW,mg/g
RV/BW,mg/g
MyocyteDiameter, �m
S 245.8�9.4 2.5�0.06 0.59�0.04 11�1.1
T1 232.2�6.4 2.8�0.14* 0.68�0.06* 13.2�1.3*
T2 234.7�17.3 3.2�0.12†‡ 0.80�0.08†‡ 14.4�1.3*
Values are mean�SD. BW indicates body weight; LV, left ventricular weight;RW, right ventricular weight.
Significant difference vs *S, †T1, P�0.05; ‡S, P�0.01.
Fernandes et al Aerobic Exercise on MicroRNA and RAS 183
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from

with the S group. Plasma renin activity also was increased by20% in T1 (P�NS) and 126% in T2 (P�0.01) whencompared with the S group (Figure 1A). In contrast, there wasa reduction in local cardiac ACE activity of 11% (P�NS) and15% (P�NS) in the RV and LV, respectively, in the T1group. When the T2 group was compared with the S group,there was a decrease of 40% (P�0.05) and 32% (P�0.05) inRV and LV, respectively (Figure 1B). Interestingly, Figure1B also shows that LV ACE2 activity was increased by 12%in T1 (1708�354 unit of fluorescence/min per milligram;P�NS) and 41% in T2 (2160�218 unit of fluorescence/minper milligram; P�0.01) when compared with the S group(1531�174 unit of fluorescence/min per milligram).
RAS Molecular AnalysisIn order to test whether swimming exercise training modu-lates cardiac RAS gene expression, real-time PCR was usedto assess ACE, ACE2, AT1, and Ang II type 2 (AT2) receptorgene expression in the heart. mRNA levels of ACE showed asmall decrease and ACE2 mRNA a small increase in bothtrained groups but without significance (data not shown). AT1
receptor gene expression increased in T1 (69%; P�0.05) andT2 (99%; P�0.01) when compared with the S group (Figure4B). In addition, AT2 receptor gene expression increased by26% (P�NS) in T1 and 332% in T2 (P�0.001); T1 differedfrom T2 (P�0.001; Figure 4D).
Similar results were obtained for RAS proteins and peptidelevels determined by Western blotting and high-performanceliquid chromatography, respectively. Figure 2A shows thatswimming exercise training decreased cardiac angiotensino-gen levels by 26% (P�0.05) in T1 and 44% in T2 (P�0.05)when compared with the S group. Because angiotensinogen isa substrate for Ang I production, this reduction was accom-panied by a decrease of 25.6% (P�0.05) in cardiac Ang Ilevels in T1 and 44% in T2 (P�0.001) when compared withthe S group (Figure 2B). The next step was to measure thelevels of ACE, because it is responsible for converting Ang Iinto Ang II. Accordingly, Figure 2C shows that cardiac ACElevels were decreased by 22% (P�NS) in T1 and 31% in T2(P�0.05) in comparison with the S group. This reduction wasaccompanied by a decrease of 23% (P�0.001) in Ang II
levels in T1 and 20% in T2 (P�0.001) in comparison with theS group (Figure 2D), indicating an attenuation of the ACE-Ang II axis induced by swimming exercise training.
Swimming exercise training also had an effect on theprotein and peptide levels of novel RAS, ACE2, and Ang(1-7) in the heart. As shown in Figure 3A, swimming trainingincreased ACE2 protein expression in both trained groups(68% in T1 and 91% in T2; P�0.05) when compared with theS group. There was increased Ang (1-7) formation (160% inT1 and 120% in T2; P�0.01; Figure 3B) in comparison withthe S group. Figure 3C shows an increase in the Ang(1-7)/Ang II ratio in both trained groups (180% in T1,P�0.05, and 160% T2, P�0.001) in comparison with the Sgroup, suggesting an aerobic training-mediated increase inAng (1-7) formation from Ang II by ACE2.
Protein expression of the AT1 receptor, in concert with theincrease in AT1 receptor mRNA levels, was 2.4-fold greaterin T1 (P�0.05) and 3.0-fold greater in T2 (P�0.05) whencompared with the S group (Figure 4A). In addition, AT2
receptor protein expression was increased 1.6-fold in T1(P�NS) and 2.2-fold (P�0.05) in T2 (Figure 4C).
MiRNAs Analysis by MicroarrayMicroarray analysis of miRNA was restricted to thosemiRNAs that underwent a significant change from baseline.Figure 5A shows miRNAs targeting ACE: in the S group therelative expression value of miRNA-27a was 1760�108arbitrary units (AU). In the T1 group the value was 2225�78AU, (26% increase in comparison with S; P�0.05), and in T2group the expression was 3218�30 AU (83% increase incomparison with S; P�0.01). In addition, T1 differed fromT2 (P�0.01). Similarly, in the S group the relative expressionvalue of miRNA-27b was 3409�89 AU. In the T1 group thevalue was 4341�124 AU, (27% increase in comparison withS; P�0.05), and in the T2 group the expression was 4939�59AU (45% increase in comparison with S; P�0.01). Inaddition, T1 differed from T2 (P�0.01). Figure 5A alsoshows miRNA targeting ACE2: in the S group the relativeexpression value of miRNA-143 was 6556�157 AU;T1�6095�83 AU was not significant when compared withS; however, in the T2 group the expression was 4249�32 AU
0
20
40
60
80
100
120
140
160
Car
diac
RA
S A
ctiv
ity(%
of c
ontr
ol)
ST1T2
†
†
0
50
100
150
200
250
Serum ACE Plasma Renin
Syst
emic
RA
S A
ctiv
ity(%
of c
ontr
ol)
*
†
†
RV ACE LV ACE LV ACE2
ST1T2
ST1T2
A B
* *
***
Figure 1. Effect of exercise training on systemic and cardiac renin-angiotensin (Ang) system (RAS) activity. Serum Ang-convertingenzyme (ACE) activity and plasma renin activity (A). Right ventricle (RV) ACE activity, left ventricle (LV) ACE activity, and LV ACE2 activ-ity (B). Groups: S indicates sedentary; T1, swimming training protocol 1; and T2, swimming training protocol 2. Data are reported asmean�SD. Significant difference vs *S, †T1, P�0.05; **S, P�0.01.
184 Hypertension August 2011
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from

(35% decrease in comparison with S; P�0.01). T1 alsodiffered from T2 (P�0.01).
MiRNAs Analysis by Real-Time PCRTo confirm the miRNAs that targeted RAS genes in physio-logical LVH, the miRNAs 27a, 27b, and 143 were quantifiedby real-time PCR. MiRNAs 27a (S: 1.0�0.08, T1:1.52�0.06, and T2: 2.04�0.13 AU) and 27b (S: 1.0�0.08,T1: 1.30�0.05, and T2: 1.59�0.08 AU) were upregulated inT1 and T2 in comparison with S, whereas miRNA-143 (S:
1.0�0.11, T1: 0.82�0.02, T2: 0.58�0.05 AU) was down-regulated in T2 in comparison with S. The miRNA expressionin T1 and T2 in comparison with the S group confirmed themicroarray results.
DiscussionThe results of the present study show that swimming exercisetraining induced physiological LVH, is not correlated withpathological cardiac hypertrophy markers, increased AT1 andAT2 receptor expression, decreased cardiac ACE and Ang II
0
0.5
1.0
1.5
2.0
2.5
S T1 T20
0.5
1.0
1.5
S T1 T2
LV A
ngio
tens
inog
en/α
-tubu
linra
tio p
rote
in le
vels
(a.u
.)
0
200
400
600
800
1000
S T1 T20
200
400
600
800
S T1 T2
B
C
D
A T1 T2S T1 T2S
ACEα- tubulin
T1 T2S T1 T2S
Angiotensinogenα- tubulin
0
*
T1 T2S T1 T2S T1 T2S T1 T2ST1 T2S T1 T2S T1 T2S T1 T2S
LV A
CE/α
-tubu
lin ra
tiopr
otei
n le
vels
(a.u
.)
LV A
ng I
leve
ls(p
mol
/g)
LV A
ng II
leve
ls(p
mol
/g)
**
****
*** ***
Figure 2. Effect of exercise training onclassic Ang-converting enzyme (ACE)-Ang II axis in the heart. Data are pre-sented as mean�SD. Cardiac angioten-sinogen (A) and ACE (C) proteinexpression analyzed by Western blottingaccompanied by their representativeblots from sedentary (S) and trainedgroups (T1 and T2). Targeted bandswere normalized for cardiac �-tubulin.Cardiac Ang I (B) and Ang II (D) peptideconcentration analyzed by high-performance liquid chromatography. Sig-nificant difference vs *S, P�0.05; ***S,P�0.001.
C
0
150
300
450
600
750
900
0
0.5
1.0
1.5
A T1 T2S T1 T2S
ACE2α- tubulin
B
S T1 T2S T1 T20
0.2
0.4
0.6
0.81.01.2
1.41.6
S T1 T2
T1 T2S T1 T2S T1 T2S T1 T2S
LV A
CE2
/α-tu
bulin
ratio
prot
ein
leve
ls (a
.u.)
LV A
ng (1
-7) l
evel
s(p
mol
/g)
LV A
ng (1
-7)/A
ng II
ratio
(a.u
.)
** **
**
******
Figure 3. Effect of exercise training on novel Ang-converting enzyme (ACE) 2-Ang (1-7) axis in the heart. Data are presented asmean�SD. Cardiac ACE2 (A) protein expression analyzed by Western blotting accompanied by its representative blot from sedentary(S) and trained groups (T1 and T2). Targeted band was normalized for cardiac �-tubulin. Cardiac Ang (1-7) peptide concentration ana-lyzed by high-performance liquid chromatography (B). Ang (1-7) generation from Ang II represented by Ang (1-7)/Ang II ratio (C). Signifi-cant difference vs *S, P�0.05; **S, P�0.01; and ***S, P�0.001.
Fernandes et al Aerobic Exercise on MicroRNA and RAS 185
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from

levels and increased ACE2 and Ang (1-7) levels, and alteredthe expression of specific miRNAs that target RAS genes.
The LV/ BW ratio, myocyte diameter, and resting brady-cardia confirmed the exercise-associated adaptations of phys-iological LVH.24,27 In contrast to markers for pathologicalhypertrophy,28 the physiological LVH reported here was notassociated with activation of fetal genes, such as atrialnatriuretic factor, skeletal muscle �-actin, and �-myosinheavy chain. There were no pathological hypertrophy mark-ers in the exercised-trained T1 or T2 groups.
The development of LVH after training did not appear toinvolve Ang II, which was decreased, supporting recentevidence from several transgenic animal models that in-creased formation of local Ang II in the heart does notdirectly develop hypertrophy, except when excess cardiacAng II enters the circulation and causes an increase in bloodpressure.12–14 Xiao et al15 reported that, in mice expressingACE only in the heart, increased cardiac Ang II was notassociated with cardiac hypertrophy. Our group demonstratedthat, in transgenic mice harboring 1, 2, 3, or 4 ACE genecopies, the magnitude of physiological LVH induced byswimming training was not correlated with ACE levels.17
Increased AT1 receptor was also found with resistancetraining.18 Losartan treatment blocked LVH in the same 2swimming training protocols.24 The mechanism for overex-pressing the AT1 receptor may be related to an independentaction of the AT1 receptor. Zou et al16 showed in vitro and invivo that AT1 receptor is a mechanical sensor and convertsmechanical stress into a biochemical signal inducing LVHwithout involvement of Ang II. Moreover, Yasuda et al29
showed that mechanical stress activates an anticlockwise
rotation of the transmembrane 7 domain of AT1 receptor,causing a conformational change of the receptor, independentof Ang II. AT1 receptors have no direct cell signalingpathway to tyrosine kinase and the mitogen-activated proteinkinase pathways for cell growth.30,31 The AT1 receptors havean indirect, membrane-transactivating step to stimulate theepidermal growth factor receptor.30,31 Inhibition of the epi-dermal growth factor receptor directly prevents Ang II–induced LVH in rats.31 The mechanical activity of exercisecould, therefore, activate the epidermal growth factor recep-tor pathway via mechanical activation of AT1, even in theabsence of Ang II.
Aerobic exercise training also increased AT2 receptorgenes and protein expression in the LV. Studies suggest thatAT1 and AT2 receptors may serve opposing functions in theheart, although they exhibit the same ligand binding affin-ity.4,12 The role of AT2 receptors in cardiac regulation is notfully understood. The AT2 receptor has been associated withdephosphorylation and inactivation of growth factor–acti-vated mitogen-activated protein kinase and inactivation ofextracellular signal–regulated kinase 1/2, providing a protec-tive role in the heart.32 Furthermore, AT2 receptor activatesNO and bradykinin, inducing vasodilation.33 Yang et al34
demonstrated that, in transgenic animals, the overexpressionof AT2 receptors preserved LV function after myocardialinfarction. The results of the present study suggest that theAT2 receptor plays a cardioprotective role in opposingdeleterious cardiac remodeling in cardiovascular disease. Incontrast to the pathological condition, an increase in AT2
receptor expression in the heart could aid vasodilatationinduced by aerobic exercise training. This modulation might
0
0.5
1.0
1.5
T1 T2ST1 T2S
B
C
D
A
0
0.5
1.0
1.5
S T1 T2
*
0
50
100
150
200
250
S T1 T2
S T1 T2
0
100
200
300
400
500
S T1 T2
‡
T1 T2S T1
AT2 receptorα- tubulin
T2ST1 T2S T1
AT1 receptorα- tubulin
T2S
LV A
T1/α
-tubu
lin ra
tiopr
otei
n le
vels
(a.u
.)
LV A
T2/α
-tubu
lin ra
tiopr
otei
n le
vels
(a.u
.)
LV A
T1 m
RN
A le
vels
(% c
ontr
ol)
LV A
T2 m
RN
A le
vels
(% c
ontr
ol)
* *
*** ***
Figure 4. Effect of exercise training onangiotensin (Ang) II type 1 (AT1) and AngII type 2 (AT2) receptor gene and proteinexpression in the heart. Data are pre-sented as mean�SD. Cardiac AT1receptor (A) and AT2 receptor (C) proteinexpression analyzed by Western blottingaccompanied by their representativeblots from sedentary (S) and trainedgroups (T1 and T2). Targeted bandswere normalized for cardiac �-tubulin.Cardiac AT1 receptor (B) and AT2 recep-tor (D) gene expression analyzed by real-time PCR. Targeted genes were normal-ized by cyclophilin mRNA. Significantdifference vs *S, P�0.05; **S, P�0.01;and ***S, ‡T1 P�0.001.
186 Hypertension August 2011
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from

increase blood and oxygen transport to the exercising cardiacmuscle to facilitate high performance.
The present study demonstrated the effect of aerobicexercise training on ACE2 and Ang (1-7) in the heart of rats.The discovery of ACE2 revealed that classic RAS has areciprocal side, the novel RAS. The results of this study showa reversal of balance in favor of the novel RAS with exercisetraining. When compared with sedentary animals, the trainedgroups had increased ACE2 and Ang (1-7) activity andprotein expression in the LV. ACE2 cleaves Ang I to generatethe inactive Ang (1-9) peptide, but the preferred pathway with500-fold greater efficiency is Ang II to generate the vasodi-lator Ang (1-7).6,35,36 In hypertension, a decrease in ACE2mRNA and protein expression, leads to increases in local AngII levels and might reduce myocardial blood flow, preferen-tially via coronary vasoconstriction or microcirculatory dys-function. However, with the use of ACE inhibitor or AT1
receptor blocker, the ACE2 level is enhanced.35–37 Transgenicanimal models overexpressing cardiac ACE2 by systemiclentiviral delivery resulted in a regression of pathologicalLVH in hypertensive rats.21 In fact, studies have suggestedthat Ang (1-7) can reduce hypertension-induced cardiac remod-
eling through a direct effect on the heart and raise the possibilitythat pathologies associated with ACE2 inactivation are partlymediated by a decrease in Ang (1-7) production.38,39
AT1 receptor blockade augmented the plasma Ang (1-7)/Ang II ratio, suggesting increased generation of Ang (1–7)from Ang II.20 In addition, Crackower et al19 showed thatdeletion of ACE2 in mice resulted in elevated cardiac andplasma Ang II, together with impaired cardiac contractilityand exhibited LV dilatation. Therefore, ACE2 might protectagainst pathological LVH by reducing Ang II concentrationand increasing Ang (1-7) generation.6,20–23,38,39
Thus, the mechanism of aerobic exercise training to preventLVH could occur by diminished vascular resistance, leading toincreased cardiac flow, attributed to the reduction in ACE andAng II levels and the vasodilator effects of the ACE2-Ang (1-7)expression, mediating the release of different vasoactive factors,such as NO, prostaglandins, and bradykinin.40,41
Another aspect of this report is the correlation of miRNAswith Ang-related genes. The implication of specific miRNA-regulating RAS genes in cardiac hypertrophy induced by exer-cise training has not been reported previously. The targetpredictions of miRNAs are all based on 3�untranslated regions ofmRNA of RAS components in Web-based bioinformatics Tar-getScan 4.2 and 5.1, MiRanda, and PicTar. Confirming thesepredictions, the literature provides more details of Ang generegulation through quantitative PCR and Ang gene measure-ments. The ACE gene has been shown recently to be regulatedby miRNA-27a and 27b.42 It has been demonstrated that theACE2 gene is regulated by miRNA-143.43
This study reveals potential molecular mechanisms for theresults. MiRNAs target multiple genes, but targeted genes arecontrolled by specific miRNAs.25,26 Increased expression ofmiRNA indicates inhibition of the target gene. This appearsto be the case with miRNA-27a and -27b, because ACEdecreased by 22% (T1) and 31% (T2) in comparison with thecontrol, whereas miRNA-27a increased by 26% (T1) and88% (T2) when compared with the control, and miRNA-27bincreased by 27% (T1) and 44% (T2). By the same principle,decreased expression of miRNAs reflects increased expres-sion of target genes. Although, in the T2 group in which theexpression of ACE2 was highest, the miRNAs that target theACE2 gene, miRNA-143, were at their lowest level of expres-sion when compared with the control or T1. Thus, aerobicexercise training exerts an effect on the expression of miRNAsand thereby might regulate their specific target genes.
PerspectivesExercise is widely recognized as an important lifestyle factorin lowering hypertension and improving cardiac health. Thisstudy reveals some of the biochemical and molecular mech-anisms of aerobic exercise training involved in physiological,nonpathological cardiac hypertrophy. The results clearlyindicate that, in aerobic exercise trained animals, LVH isphysiological and associated with decreased ACE and Ang IIversus increased ACE2 and Ang (1-7) and increased AT1 andAT2 receptors. In addition, there was a reciprocal differentialexpression of specific miRNAs and these genes. Thesefindings are consistent with a model in which exercise mayinfluence these changes, at least in part, altering the expres-
A
B
ST1T2
ST1T2
ST1T2
ST1T2
0
20
40
60
80
100
120
140
160
180
200
miRNA-27a miRNA-27b miRNA-143
†
*
§
0
50
100
150
200
250
miRNA-27a miRNA-27b miRNA-143
LV m
iRN
A L
evel
s by
Mic
roar
ray
(% o
f con
trol
)LV
miR
NA
Lev
els
by R
eal-T
ime
PCR
(% o
f con
trol
)
*
**
**
§
**
§
**
**
†
**
†
**
Figure 5. Effect of exercise training on specific microRNAs(miRNAs) targeting renin-angiotensin (Ang) system genes. MiRNAsassociated with Ang-converting enzyme (ACE; miRNA-27a and27b) and ACE2 (miRNA-143) analyzed by microarray (A). Confirma-tion of miRNAs-27a, -27b, and -143 by real-time PCR (B). Tar-geted miRNAs were normalized by U6 expression. MiRNAs wereisolated using the mirVana quantitative RT-PCR miRNA. Significantdifference vs *S, †T1, P�0.05 and **S, §T1, P�0.01.
Fernandes et al Aerobic Exercise on MicroRNA and RAS 187
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from

sion of specific miRNAs targeting RAS genes. Together,these effects might provide the additional aerobic capacityrequired by the exercised heart. The results imply that adecrease in miRNA-143 could upregulate cardioprotectivegenes in the heart, and an increase of miRNA-27 expressioninhibits ACE levels. These results suggests that a basis fortreatment to prevent of the development of pathological LVHmight be to inhibit specific miRNAs, probably with antisenseor small interfering RNA, to inhibit ACE and Ang II andincrease ACE2 and Ang (1-7).
Sources of FundingThe present investigation was supported by grants from Fundacao deAmparo a Pesquisa do Estado de Sao Paulo (No. 2009/18370-3).E.M.O. was the recipient of a Conselho Nacional de Desenvolvi-mento Científico e Technologico-Pos-Doutorado no Exterior Fel-lowship (No.200994/2007-7) and holds scholarships from ConselhoNacional de Desenvolvimento Científico e Technologico, Brazil.T.F. was the recipient of a Fundacao de Amparo a Pesquisa doEstado de Sao Paulo Fellowship (No.07/56771-4), and N.Y.H. wasthe recipient of a Coordenação de Aperfeiçoamento de Pessoal deNível Superior Fellowship. M.I.P. was supported by National Insti-tutes of Health grant 1 R01 HL 077602.
DisclosuresNone.
References1. Dorn II GW. The fuzzy logic of physiological cardiac hypertrophy.
Hypertension. 2007;49:962–970.2. Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intra-
cellular signaling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600.3. McMullen JR, Jennings GL. Differences between pathological and phys-
iological cardiac hypertrophy: novel therapeutic strategies to treat heartfailure. Clin Exp Pharmacol Physiol. 2007;34:255–262.
4. Dostal DE. The cardiac renin-angiotensin system: novel signaling mech-anisms related to cardiac growth and function. Regul Pept. 2000;91:1–11.
5. Shikata C, Takeda A, Takeda N. Effect of an ACE inhibitor and an AT1receptor antagonist on cardiac hypertrophy. Mol Cell Biochem. 2003;248:197–202.
6. Grobe JL, Mecca AP, Lingis M, Shenoy V, Bolton TA, Machado JM,Speth RC, Raizada MK, Katovich MJ. Prevention of angiotensinII-induced cardiac remodeling by angiotensin-(1–7). Am J Physiol HeartCirc Physiol. 2007;292:736–742.
7. Kang N, Walther T, Tian XL, Bohlender J, Fukamizu A, Ganten D, BaderM. Reduced hypertension-induced end-organ damage in mice lackingcardiac and renal angiotensinogen synthesis. J Mol Med. 2002;80:359–366.
8. Dahlof B, Zanchetti A, Diez J, Nicholls MG, Yu CM, Barrios V, AurupP, Smith RD, Johansson M, for the REGAAL Study Investigators. Effectsof losartan and atenolol on left ventricular mass and neurohormonalprofile in patients with essential hypertension and left ventricular hyper-trophy. J Hypertens. 2002;20:1855–1864.
9. Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additiveamelioration of left ventricular remodeling and molecular alterations bycombined aldosterone and angiotensin receptor blockade after myocardialinfarction. Cardiovasc Res. 2005;67:97–105.
10. Lal A, Veinot JP, Ganten D, Leenen FHH. Prevention of cardiacremodeling after myocardial infarction in transgenic rats deficient in brainangiotensinogen. J Mol Cell Cardiol. 2005;39:521–529.
11. Ferreira JC, Bacurau AV, Evangelista FS, Coelho MA, Oliveira EM,Casarini DE, Krieger JE, Brum PC. The role of local and systemic reninangiotensin system activation in a genetic model of sympathetichyperactivity-induced heart failure in mice. Am J Physiol Regul IntegrComp Physiol. 2008;294:26–32.
12. Reudelhuber TL, Bernstein KE, Delafontaine P. Is angiotensin II adirector mediator of left ventricular hypertrophy? Time for another look.Hypertension. 2007;49:1196–1201.
13. Van Kats JP, Methot D, Paradis P, Silversides DW, Reudelhuber TL. Useof a biological peptide pump to study chronic peptide hormone action in
transgenic mice. Direct and indirect effects of angiotensin II on the heart.J Biol Chem. 2001;276:44012–44017.
14. Xiao HD, Fuchs S, Campbell DJ, Lewis W, Dudley SC Jr, Kasi VS, HoitBD, Keshelava G, Zhao H, Capecchi Mr, Bernstein KE. Mice withcardiac-restricted angiotensin-converting enzyme (ACE) have atrialenlargement, cardiac arrhythmia, and sudden death. Am J Pathol. 2004;165:1019–1032.
15. Xiao HD, Fuchs S, Bernstein EA, Li P, Campbell DJ, Bernstein KE. Miceexpressing ACE only in the heart show that increased cardiac angiotensinII is not associated with cardiac hypertrophy. Am J Physiol Heart CircPhysiol. 2008;294:659–667.
16. Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N,Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T,Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stressactivates angiotensin II type 1 receptor without the involvement of an-giotensin II. Nat Cell Biol. 2004;6:499–506.
17. Evangelista FS, Krieger JE. Small gene effect and exercise training-induced cardiac hypertrophy in mice: an Ace gene dosage study. PhysiolGenomics. 2006;27:231–236.
18. Barauna VG, Magalhaes FC, Krieger JE, Oliveira EM. AT1 receptorparticipates in the cardiac hypertrophy induced by resistance training inrats. Am J Physiol Regul Integr Comp Physiol. 2008;295:R381–R387.
19. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE,Oliveira-dos-Santos JA, Da Costa J, Zhang L, Pei Y, Scholey J, FerrarioCM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM.Angiotensin-converting enzyme 2 is an essential regulator of heartfunction. Nature. 2002;417:822–828.
20. Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB, FerrarioCM. Upregulation of angiotensin converting enzyme-2 after myocardialinfarction by blockade of angiotensin II receptors. Hypertension. 2004;43:970–976.
21. Huentelman MJ, Grobe Jl, Vasquez J, Stewart JM, Mecca AP, KatovichMJ, Ferrario CM, Raizada MK. Protection from angiotensin II-inducedcardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2in rats. Exp Physiol. 2005;90:783–790.
22. Díez-Freire C, Vasquez J, Correa de Adjounian MF, Ferrari MF, Yuan L,Silver X, Torres R, Raizada MK. ACE2 gene transfer attenuateshypertension-linked pathophysiological changes in the SHR. PhysiolGenomics. 2006;27:12–19.
23. Raizada MK, Ferreira AJ. ACE2: a new target for cardiovascular diseasetherapeutics. J Cardiovasc Pharmacol. 2007;50:112–119.
24. Oliveira EM, Sasaki MS, Cerencio M, Barauna VG, Krieger JE. Localrenin-angiotensin system regulates left ventricular hypertrophy inducedby swimming training independent of circulating renin: a pharmaco-logical study. J Renin Angiotensin Aldosterone Syst. 2009;10:15–23.
25. Van Rooij E, Olson EN. Micro RNAs: powerful new regulators of heartdisease and provocative therapeutic agents. J Clin Invest. 2007;117:2369–2376.
26. Van Rooij E, Marshall WS, Olson EN. Toward microRNA-based thera-peutics for heart disease: the sense in antisense. Circ Res. 2008;103:919–928.
27. Medeiros A, Oliveira EM, Gianolla R, Casarini DE, Negrao CE, BrumPC. Swimming training increases cardiac vagal activity and inducescardiac hypertrophy in rats. Braz J Med Biol Res. 2004;37:1909–1917.
28. Izumo S, Nadal-Ginard BE, Mahdavi V. Protooncogene induction andreprogramming of cardiac gene expression produced by pressureoverload. Proc Natl Acad Sci U S A. 1988;85:339–343.
29. Yasuda N, Miura S, Akazawa H, Tanaka T, Qin Y, Kiya Y, Imaizumi S,Fujino M, Ito K, Zou Y, Fukuhara S, Kunimoto S, Fukuzaki K, Sato T,Ge J, Mochizuki N, Nakaya H, Saku K, Komuro I. Conformational switchof angiotensin II type 1 receptor underlying mechanical stress-inducedactivation. EMBO Rep. 2008;9:179–186.
30. Kagiyama S, Eguchi S, Frank G, Inagami T, Zhang CL, Phillips MI.Angiotensin II induced cardiac hypertrophy and hypertension areattenuated by epidermal growth factor receptor antisense. Circulation.2002;106:909–912.
31. Kagiyama S, Qian K, Kagiyama T, Phillips MI. Antisense to epidermalgrowth factor receptor prevents the development of left ventricular hy-pertrophy. Hypertension. 2003;41:824–829.
32. Horiuchi M, Akishita M, Dzau VJ. Recent progress in angiotensin II type2 receptor research in the cardiovascular system. Hypertension. 1999;33:613–621.
33. Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S,Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, Miwa T, Kawada N,Mori Y, Shibasaki Y, Tanaka Y, Fujiyama S, Koyama Y, Fujiyama A,
188 Hypertension August 2011
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Takahashi H, Iwasaka T. Angiotensin II type 2 receptor overexpressionactivates the vascular kinin system and causes vasodilation. J Clin Invest.1999;104:925–935.
34. Yang Z, Bove CM, French BA, Epstein FH, Berr SS, DiMaria JM,Gibson JJ, Carey RM, Kramer CM. Angiotensin II type 2 receptoroverexpression preserves left ventricular function after myocardialinfarction. Circulation. 2002;106:106–111.
35. Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, TallantEA, Diz DI, Gallagher PE. Effect of angiotensin-converting enzymeinhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–2610.
36. Chappell MC. Emerging evidence for a functional angiotensin-convertingenzyme 2-angiotensin-(1–7)-Mas receptor axis: more than regulation ofblood pressure? Hypertension. 2007;50:596–599.
37. Ferrario CM. Angiotensin-converting enzyme 2 and angiotensin-(1–7): anevolving story in cardiovascular regulation. Hypertension. 2006;47:515–521.
38. Mercure C, Yogi A, Callera GE, Aranha AB, Bader M, Ferreira AJ,Santos RA, Walther T, Touyz RM, Reudelhuber TL. Angiotensin(1–7)blunts hypertensive cardiac remodeling by a direct effect on the heart.Circ Res. 2008;103:1319–1326.
39. Ferreira AJ, Castro CH, Guatimosim S, Almeida PW, Gomes ER, Dias-Peixoto MF, Alves MN, Fagundes-Moura CR, Rentzsch B, Gava E,Almeida AP, Guimaraes AM, Kitten GT, Reudelhuber T, Bader M,Santos RA. Attenuation of isoproterenol-induced cardiac fibrosis intransgenic rats harboring an angiotensin-(1–7)-producing fusion proteinin the heart. Ther Adv Cardiovasc Dis. 2010;4:83–96.
40. Li P, Chappell MC, Ferrario CM, Brosnihan KB. Angiotensin-(1–7)augments bradykinin-induced vasodilation by competing with ACE andrealizing nitric oxide. Hypertension. 1997;29:394–400.
41. Carvalho MB, Duarte FV, Faria-Silva R, Fauler B, da Mata Machado LT,de Paula RD, Campagnole-Santos MJ, Santos RA. Evidence for Mas-mediated bradykinin potentiation by the angiotensin-(1–7) nonpeptidemimic AVE 0991 in normotensive rats. Hypertension. 2007;50:762–767.
42. Goyal R, Goyal D, Leitzke A, Gheorghe CP, Longo LD. Brain renin-an-giotensin system: fetal epigenetic programming by maternal proteinrestriction during pregnancy. Reprod Sci. 2010;17:227–238.
43. Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, Braun T.Acquisition of the contractile phenotype by murine arterial smoothmuscle cells depends on the Mir143/145 gene cluster. J Clin Invest.2009;119:2634–2647.
Fernandes et al Aerobic Exercise on MicroRNA and RAS 189
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Casarini, Adriana K. Carmona, José E. Krieger, M. Ian Phillips and Edilamar M. OliveiraTiago Fernandes, Nara Y. Hashimoto, Flávio C. Magalhães, Fernanda B. Fernandes, Dulce E.
Regulation of Angiotensin-Converting Enzyme 2-Angiotensin (1-7)MicroRNAs, Decreased Angiotensin-Converting Enzyme-Angiotensin II, and Synergistic
Induced Left Ventricular Hypertrophy Involves Regulatory−Aerobic Exercise Training
Print ISSN: 0194-911X. Online ISSN: 1524-4563 Copyright © 2011 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Hypertension doi: 10.1161/HYPERTENSIONAHA.110.168252
2011;58:182-189; originally published online June 27, 2011;Hypertension.
http://hyper.ahajournals.org/content/58/2/182World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://hyper.ahajournals.org/content/suppl/2011/06/24/HYPERTENSIONAHA.110.168252.DC1Data Supplement (unedited) at:
http://hyper.ahajournals.org//subscriptions/
is online at: Hypertension Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialHypertensionin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on July 16, 2018http://hyper.ahajournals.org/
Dow
nloaded from

ONLINE DATA SUPPLEMENT
Aerobic exercise training induced LVH involves regulatory microRNAs, decreased ACE-
ANG II, and synergistic regulation of ACE2-ANG (1-7).
Short title: Effect of aerobic exercise training on cardiac RAS.
Tiago Fernandes, MD1, Nara Y. Hashimoto, MD1, Flávio C. Magalhães, MD1, Fernanda B.
Fernandes, MD2, Dulce E. Casarini, PhD2, Adriana K. Carmona, PhD3, José E. Krieger, PhD4,
Michael I. Phillips PhD5, Edilamar M. Oliveira, PhD1,5.
1Laboratory of Biochemistry of the Motor Activity, School of Physical Education and Sport,
University of Sao Paulo, SP, Brazil. 2Nephrology Division, Kidney and Hypertension Hospital, UNIFESP, SP, Brazil.
3Department of Biophysics, UNIFESP, SP, Brazil. 4Laboratory of Genetics and Molecular Cardiology, InCor, Medical School,
University of Sao Paulo, SP, Brazil. 5Laboratory of Stem Cells, Keck Graduate Institute, Claremont, CA, USA.
Address for correspondence:
Edilamar Menezes de Oliveira, PhD
School of Physical Education and Sport, University of Sao Paulo.
Laboratory of Biochemistry of the Motor Activity.
Av. Professor Mello Moraes, 65- Cidade Universitária- São Paulo- SP
05508-900-Brazil
Phone: (5511) 3091-2118, Fax: (5511) 3813-5921
E-mail: [email protected]

DETAILED METHODS Exercise Training Protocols
Protocol 1 (T1) low intensity, moderate volume exercise: training consisted of swimming sessions of 60-minute duration, 5 days a week, for 10 weeks, which were carried out between 11:30 AM and 1:30 PM. Protocol 2 (T2) low intensity, high volume exercise: Animals assigned to T2 performed the same swimming training protocol as T1 until the end of the 8th week. On the 9th week rats trained twice a day, swimming sessions of sixty-minute duration with four hours interval between sessions. In that week swimming sessions were carried on from 07:30 to 08:30 AM and from 12:30 AM to 1:30 PM. On the 10th week rats trained three times a day, with sessions of sixty-minute duration with four hours interval between sessions. In that week swimming sessions were carried on from 07:30 to 08:30 AM, from 12:30 AM to 1:30 PM, and from 5:30 to 6:30 PM. The aim of increasing training frequency (T2) was to induce the magnitude of cardiac hypertrophy.
Animals were trained in a swimming apparatus specially designed to allow individual exercise training of rats. The system consisted of two coupled 700 L water glass tanks of different dimensions. The outer tank measures 60 cm in diameter, 1.60 cm in width and 90 cm in height. The inner tank is divided into 14 lanes with a surface area of 20 x 20 cm per lane and a depth of 60 cm. A heating system kept the water temperature between 30-32oC and a water filter with a flow capacity of 420 L.h-1 was used to clean the swimming apparatus (1).
It was showed in our previous study that this swimming-training protocol did not stress the animals as it did not alter plasma catecholamine concentration (2). Exercise duration and workload were increased gradually until rats could swim for 60 minutes wearing caudal dumbbells weighing 5% of their body weight. Thereafter, duration and dumbbells were kept constant. All animals were weighed once a week and the workload adjusted to body weight variations. Sedentary groups were placed in the swimming apparatus for 10 minutes twice a week without workload to control for being in the water. O2 uptake of rats swimming individually is about 50-65% of maximum oxygen uptake. This low intensity long period training protocol is effective for promotion of cardiovascular adaptations and for increase of muscle oxidative capacity. These protocols have already been used previously in our laboratory (1,2). Hemodynamic Parameters: Blood Pressure (BP) and Heart Rate (HR)
Twenty-four hours after the last training session BP and HR were recorded. Rats were anesthetized (Ketamine 90 mg.kg-1 and Xylazine 10 mg.kg-1, i.p.) and a cannula (PE-50) was inserted into the carotid artery and emerged through the back of the neck. Twenty-four hours after the cannula was implanted, this cannula was connected to a strain-gauge transducer (P23 Db; Gould-Statham). Arterial blood pressure was recorded on a beat to beat basis (AT/CODAS) at a frequency of 1000Hz for 30 min in quiet, conscious, unrestrained rats. Recorded data indicated the average of all values of systolic, diastolic, heart rate and mean arterial pressure over the entire recording time of 30 min.
Samples preparation
At the end of the experimental period the rats were decapitated and blood and tissue samples (heart) were harvested, weighed, frozen, stored at -80o C and used within 1 month for enzymes assay, miRNA, mRNA and protein preparation. To determine plasma

angiotensin II the first 3 ml of trunk blood (a mixture of venous and arterial blood) was rapidly collected in chilled glass tubes containing a mixture of potassium EDTA (25 mmol), o-phenanthroline (0.44 mmol), pepstatin A (0.12 mmol), and 4-chloromercuribenzoic acid (1 mmol). This mixture of protease inhibitors prevented the in vitro production and degradation of angiotensin peptides (3). To determine the renin activity the blood was collected with EDTA (25 mmol). The blood was centrifuged the plasma was separated and stored at -20oC.
Measurement of Cardiac Hypertrophy
To measure cardiac mass, the hearts were stopped at diastole by perfusion of 14 mM KCl. After the heart was weighed, the left ventricle (LV) was dissected corresponding to the remaining tissue upon removal of both atria and the free wall of the right ventricle (RV). The interventricular septum remained as part of the LV. Cardiac hypertrophy was assessed by the measurement of the ratio of LV and RV weight in milligrams to animal body weight (BW) in grams (LV/BW and RV/BW in mg.g-1). The LV was fixed in 6% formaldehyde and embedded in paraffin, cut in 5 μm sections, from the level of papillary muscle and subsequently stained with hematoxylin and eosin (HE) for the visualization of cellular structures. Two randomly selected sections from each animal were visualized by light microscopy using an oil immersion objective with a calibrated magnification (x400). Myocytes with visible nucleus and intact cellular membranes were chosen for diameter determination. The width of individually isolated cardiomyocytes were displayed on a viewing screen that was manually traced, across the middle of the nuclei, with a digitizing pad and determined by a computer assisted image analysis system (Quantimet 520; Cambridge Instruments). For each animal, approximately 20 visual fields were analyzed.
RAS Biochemical Analysis Cardiac and Serum angiotensin-converting enzyme (ACE) activity:
ACE activity was determined in heart tissue and serum using Abz-FRK(Dnp)P-OH derivatives as substrates by continuously measuring the fluorescence according to Alves et al. (4). Heart samples were quickly harvested, homogenized in 0.1 M Tris-HCl buffer, pH 7.0, containing 50 mM NaCl and centrifuged at 1000 g for 10 min. The assays were performed at 37ºC in 0.1 M Tris-HCl buffer, pH 7.0, containing 50 mM NaCl and 10 µM ZnCl2. The hydrolysis rate of the intramolecularly quenched fluorogenic substrate Abz-FRK-(Dnp)P-OH (10 uM) incubated with aliquots of heart homogenate and serum for 30 min at 37ºC was assessed to obtain ACE enzymatic activity. Fluorescence increments along the time were read at 420nm emission: 320nm excitation. Heart and serum ACE activity were expressed as UF.min-1.mg-1of protein x 1000. The protein content was determined by the Bradford methods (5) by using bovine serum albumin as the standard (Bio-Rad Protein Assay). Cardiac angiotensin-converting enzyme 2 (ACE2) activity:
ACE2 activity was determined in heart tissue by the same method described above. However, this method uses a fluorescent peptide Abz-APK(Dnp)-OH in 0.2 M Tris-HCl buffer, 200 mM NaCl, 2 µg BSA, pH 7.5 which is hydrolyzed with high affinity by ACE2 (Kcat/Km = 3,5 x 104 M-1.s-1). ACE2 activity was expressed in UF.min-1.mg-1of protein. Plasma renin activity assay:
Plasma renin activity (PRA) was determined by angiotensin radioimmunoassay, using a commercial kit (REN-CT2, CIS Bio International, Gif-sur-Yvette, France). This

assay allows direct measurement of PRA. Results were quantified in a Gama Counter, and the enzyme activity was expressed as ng Ang I/mL/h.
MiRNAs and RAS Molecular Analysis RNA extraction and MiRNA Microarray
Frozen LV samples (100 mg) were homogenized in Trizol and RNA was isolated according to the manufacture (Invitrogen Life Technologies, CA, USA). Following extraction, the RNA total concentration was quantified using NanoDrop Specthophotometer (Nano-Drop Technologies, USA) and checked for integrity by EtBr-agarose gel electrophoresis. MiRNA was isolated using the mirVanaTM qRT-PCR- miRNA Isolation Kit (Ambion, TX, USA). RNA from two animals in each group was pooled and used for miRNA expression analysis (LC Science, TX, USA) with the Agilent platform. The arrays consist of 15,000 features including rat probes for 349 miRNAs based on Sanger miRBASE 13.0. The Agilent miRNA platform requires 100ng of total RNA per labelling reaction. The quality of all RNA samples was checked using the miRMAX microarray. Results are expressed as arbitrary units (a.u.). TargetScan program was used to search the miRNAs target to the RAS. Real-Time Reverse Transcriptase-Polymerase Chain Reaction:
The relative gene expression of α-MHC (alfa-Myosin Heavy Chain), β-MHC (beta-Myosin Heavy Chain), ANP (Atrial Natriuretic Peptide), skeletal α-actin, ACE, ACE2, AT1a, and AT2 mRNA in the LV was analyzed by real-time PCR. In addition, miRNAs- 27a, 27b and 143 also were quantified by real-time PCR. cDNA synthesis
RNA were primed with 0.5 µg/µl oligo dT (12–18 bp) (Invitrogen Life Technologies, CA, USA) to generate the first strand DNA. Reverse transcription (RT) was performed using SuperScriptTM II Reverse Transcriptase (Invitrogen Life Technologies, CA, USA).
cDNA for miRNA analysis was synthesized from total RNA using gene-specific primers according to the TaqMan MicroRNA Assay protocol (Applied Biosystems, CA, USA). The 15 μl reactions obtained by TaqMan MicroRNA Reverse Transcription Kit protocol (Applied Biosystems, CA, USA) were incubated in Thermal Cycler for 30 min at 16°C, 30 min at 42°C, 5 min at 85°C and then held at 4°C. Real-Time PCR:
Prior to analyzing samples, a standard curve for each amplicon was obtained using serial dilutions of cDNA to determine amplification primer efficiency and the amount of material for each reaction. Primers were designed using Primer 3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3 www.cgi). DNA sequence was obtained from GenBank and primers were made in separate exons to distinguish by size PCR products derived from cDNA from those derived from genomic DNA contaminants. The mRNA expression were assessed by oligonucleotides primers as follows: for α-MHC: (sense: 5’-CGA GTC CCA GGT CAA CAA G-3’, antisense: 5’-AGG CTC TTT CTG CTG GAC C-3’); for β-MHC: (sense: 5’-CAT CCC CAA TGA GAC GAA G-3’, antisense: 5’-AGG CTC TTT CTG CTG GAC A-3’); for ANP: (sense: 5’- CTT CGG GGG TAG GAT TGA C-3’, antisense: 5’-CTT GGG ATC TTT TGC GAT CT-3’); for skeletal α-actin: (sense: 5’-ACC ACA GGC ATT GTT CTG GA-3’, antisense: 5’-TAA GGT AGT CAG TGA GGT CC-3’); for ACE: (sense: 5’ CTG CCT CCC AAC GAG TTA GAA 3’, antisense: 5’-CGG GAC GTG GCC ATT ATA TT 3’); for ACE2: (sense: 5’ CAT

TGG AGC AAG TGT TGG ATC TT 3’, antisense: 5’ GAG CTA ATG CAT GCC ATT CTC A 3’); for AT1a (sense: 5’-CAC AAC CCT CCC AGA AAG TG-3’, antisense: 5’-AGG GCC ATT TTG TTT TTC TG-3’) and for AT2 (sense: 5’- GGC CTG TTT GTC CTC ATT GC -3’, antisense: 5’- CAC GGG TTA TCC TGT TCT TC -3’).
Real-time PCR quantification of the target mRNAs was performed with a SYBR Green PCR Master Mix, (Applied Biosystem, CA, USA) using ABI PRISM 7700 Sequence Detection System (Applied Biosystem, CA, USA). The expression of cyclophilin (sense: 5’-AAT GCT GGA CCA AAC ACA AA -3’, antisense: 5’-CCT TCT TTC ACC TTC CCA AA -3’) was measured as an internal control for sample variation in RT reaction. An aliquot of the RT reaction was used for 50 cycle PCR amplification in the presence of SYBR green fluorescent dye according to a protocol provided by the manufacturer (Applied Biosystems, CA, USA).
In order to accurately detect mature miRNAs and confirm array results, real-time PCR quantification was performed using TaqMan MicroRNA Assay protocol (Applied Biosystems, CA, USA). The 20 μl PCR included 1.33 μl RT product, 10 μl TaqMan Universal PCR master mix II (2×), 7.67 μl nuclease-free water and 1 μl of primers and probe mix of the TaqMan MicroRNA Assay protocol for miRNAs- 27a (INV 408) , 27b (INV 409) and 143 (INV 466). The reactions were incubated in a 96-well optical plate at 95°C for 10 min, followed by 40 cycles of 95°C for 15s and 60° for 1 min. Samples were normalized by evaluating U6 expression.
PCR product generation was monitored by measuring the increase in fluorescence caused by the binding of SYBR green to double-stranded DNA or by the probe presence in TaqMan MicroRNA Assay at each annealing phase. A dissociation curve observed in SYBR green analysis was generated at the end of the reaction to verify that a single product was amplified. Each heart sample was analyzed in triplicate. Relative quantities of target gene expressions of sedentary rats vs. trained rats were compared after normalization to the values of internal control (ΔCT). Fold change in mRNA expression were calculated using the differences in ΔCT values between the two samples (ΔΔCT) and equation 2-ΔΔCT. Results are expressed as % of control. Western Blotting analysis
The protein expression of angiotensinogen, ACE, ACE2, AT1 and AT2 receptors in the left ventricle was analyzed by western blotting. The frozen ventricles (100 mg) were homogenized in cell lyses buffer containing 100 mM Tris-HCl, 50 mM NaCl, 1% Triton X-100 and protease inhibitor cocktail (1:100, Sigma-Aldrich, MO, USA). Insoluble heart tissues were removed by centrifugation at 10,000 × g, 4° C, 10 min. Samples were loaded and subjected to SDS-PAGE in 8% polyacrylamide gels. After electrophoresis, proteins were electro-transferred to nitrocellulose membrane (Amersham Biosciences, NJ, USA). Equal loading of samples (50 µg) and even transfer efficiency were monitored with the use of 0.5% Ponceau S staining of the blot membrane. The blot membrane was then incubated in a blocking buffer (5% nonfat dry milk, 10mM Tris-HCl, pH 7.6, 150 mM NaCl, and 0.1% Tween 20) for 2h at room temperature and then incubated overnight at 4ºC with mouse anti-AT1 receptor monoclonal antibody and rabbit anti-AT2 receptor polyclonal antibody (1:1000 and 1:800, respectively; Abcam, Cambridge, UK), mouse anti-ACE clone 2E2 monoclonal antibody (1:1000, Chemicon International, CA, USA), goat anti-ACE2 and anti-Ang I/II precursor polyclonal antibody (1:1000 and 1:500, respectively; Santa Cruz Biotechnology Inc., CA, USA). Binding of the primary antibody was detected with

the use of peroxidase-conjugated secondary antibodies and enhanced chemiluminescence reagents (Amersham Biosciences, NJ, USA) were used to visualize the autoradiogram, which was later exposed to photographic film. The film was developed and the bands were analyzed using Scion Image software (Scion Corporation based on NIH image). Cardiac α-tubulin expression levels were used to normalize the results. Results are expressed as arbitrary units (a.u.).
Angiotensin I, II and (1-7) Quantification by High Performance Liquid Chromatography (HPLC)
Left ventricle was weighed and homogenized in 100 mM sodium phosphate buffer pH 7.2, 340 mM sucrose and 300 mM NaCl, containing protease inhibitor cocktail (1:100, Sigma-Aldrich, MO, USA). The samples were centrifuged at 10,000 × g, 4° C, 20 min.
The extraction of angiotensins was held in Oasis C18 columns (Waters, MA, USA) previously activated with methanol (5 mL), tetrahydrofuran (5 mL), hexane (5 mL), methanol (5 mL) and water (10 mL). After activation, the samples were applied to the columns, washed with water and eluted in ethanol / acetic acid / water in the proportion 90% / 4% / 6%. The eluted fractions were lyophilized and resuspended in 500 uL of mobile phase A (5% acetonitrile in 0.1% orthophosphoric acid) and filtered with 0.22 mm membrane for analysis by high-performance liquid chromatography (HPLC, Shimadzu System, Japan).
The angiotensin of each sample were separated on a reversed phase column ODS Aquapor 300 (250 x 4.6 mm), 7μ (PerkinElmer’s Browlee Columns) using the gradient from 5–35% of mobile phase B (95% acetonitrile in 0.1% phosphoric acid) under a flow of 1.5 mL / min for 40 min. The angiotensins were identified by comparing them with the retention time of standard angiotensins. Results were expressed as pmol/g of tissue. Statistical analysis
Results are represented as mean ± SD. Statistical analysis was performed using one-way ANOVA. P values <0.05 were accepted as statistically significant. Tukey’s post hoc test was used for individual comparisons between means when a significant change was observed with ANOVA.
REFERENCES: 1. Oliveira EM, Sasaki MS, Cerêncio M, Baraúna VG, Krieger JE. Local renin-angiotensin system regulates left ventricular hypertrophy induced by swimming training independent of circulating renin: a pharmacological study. J Renin Angiotensin Aldosterone Syst. 2009; 10:15-23. 2. Medeiros A, Oliveira EM, Gianolla R, Casarini DE, Negrão CE, Brum PC. Swimming training increases cardiac vagal activity and induces cardiac hypertrophy in rats. Braz J Med Biol Res. 2004; 37: 1909-1917. 3. Kohara K, Tabuchi Y, Senanayake P, Brosnihan KB, Ferrario CM. Reassessment of plasma angiotensins measurement: effects of protease inhibitors and sample handling procedures. Peptides. 1991; 12: 1135–1141.

4. Alves MFM, Araujo MC, Juliano, MA, Oliveira EM, Krieger JE, Casarini DE, Juliano L, Carmona AK. A continuous fluorescent assay for the determination of plasma and tissue angiotensin I-converting enzyme activity. Braz J Med Biol Res. 2005, 38:861-868. 5. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976. 72: 248-254.
Supplementary Figure Legend Figure S1. Effect of swimming exercise training on the cardiomyocytes diameter (μm). Representative histological sections of LV myocytes diameter in the sedentary (A), T1- swimming training protocol 1 (B) and T2- swimming training protocol 2 (C). The arrows indicate the lines showing the width of individually isolated cardiomyocytes that was manually traced, across the middle of the nuclei, visualized by light microscopy (x400). Significant difference vs. *S, P<0.05.
A CB