acquired resistance to bet-protacs(proteolysis targeting ... · preclinical models of leukemia,...
TRANSCRIPT

1
Acquired Resistance to BET- PROTACs (Proteolysis Targeting Chimeras) Caused by Genomic Alterations
in Core Components of E3 ligase Complexes
Lu Zhang1,*, Bridget Riley-Gillis2, Priyanka Vijay2, Yu Shen1,*
1 Oncology Discovery, AbbVie Inc., North Chicago, Illinois
2 Genomic Research Center, AbbVie Inc., North Chicago, Illinois
*Co-corresponding Authors:
Lu Zhang, AbbVie Inc., 1 North Waukegan Road, North Chicago, IL, 60064. Phone: 8479382212;
Email:[email protected] and
Yu Shen, AbbVie Inc., 1 North Waukegan Road, North Chicago, IL, 60064. Phone: 8479361128;
Email:[email protected]
Running title: Defects in E3 ligase complex cause resistance to BET-PROTACs
Key words: BET-PROTACs; drug resistance; E3 ligase
Disclosure of Potential Conflicts of Interest
All authors are employees of AbbVie. The design, study conduct, and financial support for this research
were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the
publication.
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

2
Abstract
Proteolysis Targeting Chimeras (PROTACs) are bifunctional molecules that hijack endogenous E3
ubiquitin ligases to induce ubiquitination and subsequent degradation of protein of interest. Recently,
it has been shown that PROTACs with robust in vitro and in vivo activities and, in some cases, drug-like
pharmaceutical properties can be generated using small molecule ligands for the E3 ligases VHL and
CRBN. These findings stoked tremendous enthusiasm on using PROTACs for therapeutics development.
Innate and acquired drug resistance often underlies therapeutic failures, particularly for cancer therapy.
With the PROTAC technology progressing rapidly towards therapeutic applications, it would be
important to understand whether and how resistance to these novel agents may emerge. Using BET-
PROTACs as a model system, we demonstrate that resistance to both VHL- and CRBN-based PROTACs
can occur in cancer cells following chronic treatment. However, unlike what was often observed for
many targeted therapeutics, resistance to BET-PROTACs did not result from secondary mutations that
affect compound binding to the target. In contrast, acquired resistance to both VHL- and CRBN-based
BET-PROTACs was primarily caused by genomic alterations that compromise core components of the
relevant E3 ligase complexes.
Introduction
Ubiquitination mediated proteolysis is a central mechanism of protein homeostasis in cells. E3
ubiquitin ligases bind to defined substrates and mediate ubiquitination and degradation of these
proteins. Cullin-RING ubiquitin ligases (CRL) are the largest family of E3 ubiquitin ligases. CRLs are
multicomponent complexes that minimally consist of a cullin, a RING finger protein, and a substrate
recognition subunit (1). Proteolysis Targeting Chimeras (PROTACs) are bifunctional molecules that hijack
endogenous E3 ubiquitin ligase to cause ubiquitination and subsequently degradation of proteins of
interest (2). VHL and Cereblon (CRBN) are the substrate recognition subunit of the cullin2-containing
VHL CRL complex and the cullin4-containing CRBN CRL complex respectively (3,4). Recently, it has been
shown that PROTACs with robust in vitro and in vivo activities and, in some cases, drug-like
pharmaceutical properties can be generated using small molecule ligands for the E3 ligases VHL and
CRBN (5-9). PROTACs targeting the bromodomain and extraterminal domain proteins (BET-PROTACs) are
the prototype of these small molecule PROTACs (5-7). BET-PROTACs trigger rapid and prolonged
degradation of BET proteins with exceptional potencies, and exhibit robust anti-tumor activities in
preclinical models of leukemia, lymphoma, prostate cancer, and triple negative breast cancers
(5,6,10,11).
Compared to traditional small molecule inhibitors, PROTACs offer several advantages. For
example, PROTACs can exert more rapid, potent, and durable inhibition of targets, abolish the
scaffolding function of a protein, and turn a non-functional binder into functional degraders. In
addition, the stringent conformational requirement and potential linker interaction between target
protein and E3 ligase within ternary complex may allow PROTACs to offer an additional layer of
selectivity over small molecule inhibitors (12-15). These unique properties make PROTACs a promising
modality for the development of next generation therapeutics. However, innate and acquired drug
resistance is a common cause of therapeutic failure, particularly for cancer therapy. With the rapid
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

3
advancement of the PROTAC technology towards therapeutic applications, it would be important to
understand whether and how drug resistance to these novel agents may emerge. In this study, using
BET-PROTACs with both VHL and CRBN ligands as a model system, we interrogate potential mechanisms
of acquired resistance to PROTACs in cancer cell lines.
Materials and Methods
Cells, compounds and antibodies
SKM1, MV4:11, LNCaP and OVCAR8 cell lines were purchased from ATCC or DSMZ and maintained by a
Core Cell Line Facility. All cell lines were tested for mycoplasma using MycoAlert Detection Kit (Lonza)
and authenticated using the Gene Print10 STR Kit (Promega) and maintained in RPMI1640 medium with
10% FBS (Gibco). ABBV-075, ARV-771 and ARV-825 were synthesized at AbbVie by the methods
according to (5,6,16). All antibodies were purchased from commercial sources as follows: antibodies
against BRD2/3/4 from Bethyl; antibodies against c-MYC, PARP, and VHL from Cell Signaling Technology;
antibody against CRBN from Thermo Fisher; antibody against CUL2 from Abcam; antibody against β-
actin from Sigma. O1R and O3R overexpressing cells were created by infection with pLOC or pLOC-CUL2
or pLOC-CRBN lentiviral particle (Dharmacon) in the presence of 10 µg/mL of polybrene. Cells were
selected with 10 µg/mL of blasticidin. Western blot analysis was performed to confirm expression of
these proteins in the cells.
Cell viability and caspase 3/7 activity assay
Cells were seeded in 96-well plates and incubated at 37°C in an atmosphere of 5% CO2. Compounds
were added at a series of dilution after overnight incubation. After 3 days incubation, Caspase-Glo3/7
luminescent and CellTiter-Glo assay (Promega) were performed according to manufacturer's instruction.
Luminescence signal from each well was measured using the Enspire luminometer (PerkinElmer), and
the data were analyzed using the GraphPad Prism software (Graph-Pad Software Inc.).
Quantitative PCR (qPCR)
Total RNA was harvested using the RNeasy Plus Mini Kit (Qiagen) and cDNA was made with the High-Capacity cDNA Reverse Transcription (RT) Kit (Applied Biosystems). qPCR was performed with TaqMan Universal Master Mix and Pr obes (Life Technologies), and analyzed using the DDCt method. Data are presented as mean ± SD. The difference between two groups was evaluated using the two-tailed Student’s t-test. P-values less than 0.05 were considered statistically significant. Western blot analysis
Cell lysates were prepared in Laemnlli buffer (Biorad). Thirty micrograms of total protein was resolved
on a 7-12% SDS polyacrylamide gel and probed with corresponding primary antibodies.
Whole exome sequencing
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

4
Samples were prepared in accordance with the manufacturer’s instructions for the KAPA Hyper Prep and
Nimblegen SeqCap EZ MedExome kits. Briefly, 100ng of DNA was sheared using a Covaris M220
sonicator (adaptive focused acoustics). DNA fragments were end-repaired, adenylated, ligated with
KAPA dual indexing adapters, and amplified by 11 cycles of PCR with the KAPA Hyper Prep kit. Samples
were pooled into a pre-capture library and targeted capture was performed using the Nimblegen
SeqCap EZ MedExome capture probe set. Captured libraries were then enriched by 14 cycles of PCR and
final libraries were evaluated using Qubit (ThermoFisher) and Agilent Tape Station and were sequenced
on an Illumina NextSeq sequencer using 2x75bp read length. The raw sequencing data is deposited at
SRA (Submission number SUB5567117).
Exome sequencing reads were processed through the Sentieon TNscope pipeline (Sentieon Inc.) for
alignment and somatic variant calling (Parental sample designated at “normal”, O1R and O3R designated
as “tumor”). Quality of sequencing data was assessed using Picard
(http://broadinstitute.github.io/picard) and MultiQC (17). SNV and indel variant calls were imported into
the VarSeq software (Golden Helix) for filtering and annotation.
Somatic copy number calling was performed using VarScan v2.4.2 using tumor/normal mpileup from
samtools v1.7 as input (18). Parameters used were minimum segment size of 100bp, minimum coverage
of 20 reads, minimum mapping quality of 20, and minimum base quality of 20. Loss-of-heterozygosity in
deleted regions were also visually confirmed by plotting minor allele frequencies from germline variant
calling with Sentieon joint HaplotypeCaller (SentieonInc.).
RNA sequencing
Each cell line was sampled in triplicate and mRNA library preparation from total RNA was conducted
following the manufacturer’s protocol for the Illumina TruSeq mRNA Preparation Kit. Briefly, 1
microgram of total RNA was purified by using poly-T oligos attached to magnetic beads then fragmented
by divalent cations under elevated temperature. The fragmented RNA underwent first strand synthesis
using reverse transcriptase and random primers. Second strand synthesis created the cDNA fragments
using DNA polymerase I and RNaseH. The cDNA fragments then went through end repair, adenylation
of the 3’ ends, and ligation of adapters. The cDNA library was enriched using 15 cycles of PCR and
purified. Final libraries were assessed using the Agilent Bioanalyzer and Qubit (ThermoFisher) assay
methods then sequenced on an Illumina NextSeq sequencer using 2x75bp read length. The raw
sequencing data is deposited at SRA (Submission number SUB5567117).
RNA sequencing reads were mapped to the human reference genome (GRCh38) using STAR aligner and
genes were quantified using featureCounts for all genes annotated in Gencode v28 (19,20). Quality of
sequencing data was assessed using Picard (http://broadinstitute.github.io/picard) and MultiQC (17).
Genes with counts per million less than 1 in two-thirds of samples or more were considered too lowly
expressed and excluded. Differential gene expression (DGE) analysis comparing parental vs. O1R and
parental vs. O3R (n=3 for each) was then performed using linear modeling in limma with TMM
normalization and voom transformation (21) DGE results were plotted using glimma (22). Sashimi plots
were prepared using Integrated Genome Viewer (23).
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

5
TaqMan CNV confirmation
Confirmation of exome copy number calls for CUL2 and CRBN was performed using TaqMan copy
number assays. 3 pre-designed assays each for CUL2 and CRBN (supplemental Table 1) were ordered
along with the reference assays for TERT and RNaseP genes. Experiments were performed according to
manufacturer’s instructions using 20ng DNA per reaction, 2X TaqMan Genotyping Master Mix, and both
a target (CUL2 or CRBN) and reference assay (TERT or RNaseP). Each assay combination was run in
quadruplicate for P, O1R, O3R and a no template control. Quantitative PCR was run on the ABI 7500 and
analyzed using the CopyCaller Software employing the DDCt method for normalization of copy calls
relative to the reference assay and parental samples.
Sanger Sequencing
Sequencing primers were designed using Primer3 software (version 1.1.4, http://www.sourceforge.net)
and were purchased from IDT (Coralville, IA) (Supplemental Table 2). Genomic DNA was amplified by
singleplex PCR using the FailSafe PCR System (Epicentre). Thermal cycling was performed with 40 cycles
[30 seconds at 98°C; 30 seconds at 62°C (-0.5°C each cycle); 60 seconds at 72°C], followed by 25 cycles
(30 seconds at 98°C; 30 seconds at 55°C; 60 seconds at 72°C). Amplicons were bidirectionally sequenced
using Big Dye Terminator version 1.1 technology on an ABI 3130xl system (Applied Biosystems).
Sequence analysis was performed using the Sequencher software version 5.4.6 (Gene Codes
Corporation).
Results
Chronic exposure to BET-PROTACs leads to drug resistance in cancer cell lines. Drug resistance often
arises following chronic exposure to cancer therapeutic agents. To investigate whether PROTACs are
subject to similar drug resistance issues, we exposed two AML (SKM-1 and MV4:11) and two solid tumor
(LNCaP and OVCAR8) cell lines to increasing concentrations of VHL- or CRBN-based BET-PROTACs over a
4 month period. The establishment of the resistant cell lines is illustrated in Supplementary Fig S1A.
Although all the four cell lines underwent apoptosis after BET-PROTAC treatment, SKM1, MV4:11 and
LNCaP cells were much more sensitive to these compounds compared to OVCAR8, likely due to the
strong dependency on BET proteins in AML and prostate cancer (Supplementary Fig S2) (6,24,25). While
no stable resistant clones were obtained from SKM-1, MV4:11 or LNCaP cells incubated with either of
the VHL- or CRBN-based BET-PROTACs, several resistant clones emerged from the OVCAR8 cells. These
resistant clones exhibited greater than 40x IC50 increase over the parental cell line for the VHL-based
BET-PROTAC ARV-771(6) or the CRBN-based BET-PROTAC ARV-825(5) (Fig. 1A). Withdrawing BET-
PROTAC from culture media for two months did not diminish resistance, indicating that resistance to
BET-PROTACs in these cells may result from stable genetic changes (Fig S1B). Additionally, the resistant
clones maintained similar sensitivities as the parental cell lines to the bromodomain inhibitor ABBV-075
(16), suggesting that the BET bromodomains in these cells retain the ability of binding BET inhibitors (Fig
S1C). It is noteworthy that the O1R cells, which are resistant to the VHL-based BET-PROTAC ARV-771,
remained sensitive to the CRBN-based BET-PROTAC ARV-825. Conversely, the O3R cells, which are
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

6
resistant to the CRBN-based BET-PROTAC ARV-825, remained sensitive to the VHL-based BET-PROTAC
ARV-771 (Fig. 1A). Consistent with what was observed in the proliferation assays, analysis of PARP
cleavage and caspase activation further confirmed that the O1R cells were resistant to ARV-771- but not
ARV-825-induced apoptosis, and the O3R cells were resistant to ARV-825- but not ARV-771 induced
apoptosis (Fig. 1B and C). Taken together, these results demonstrate that the O1R cells are specifically
resistant to the VHL-based BET-PROTAC (ARV-771), and the O3R cells are specifically resistant to the
CRBN-based BET-PROTAC (ARV-825).
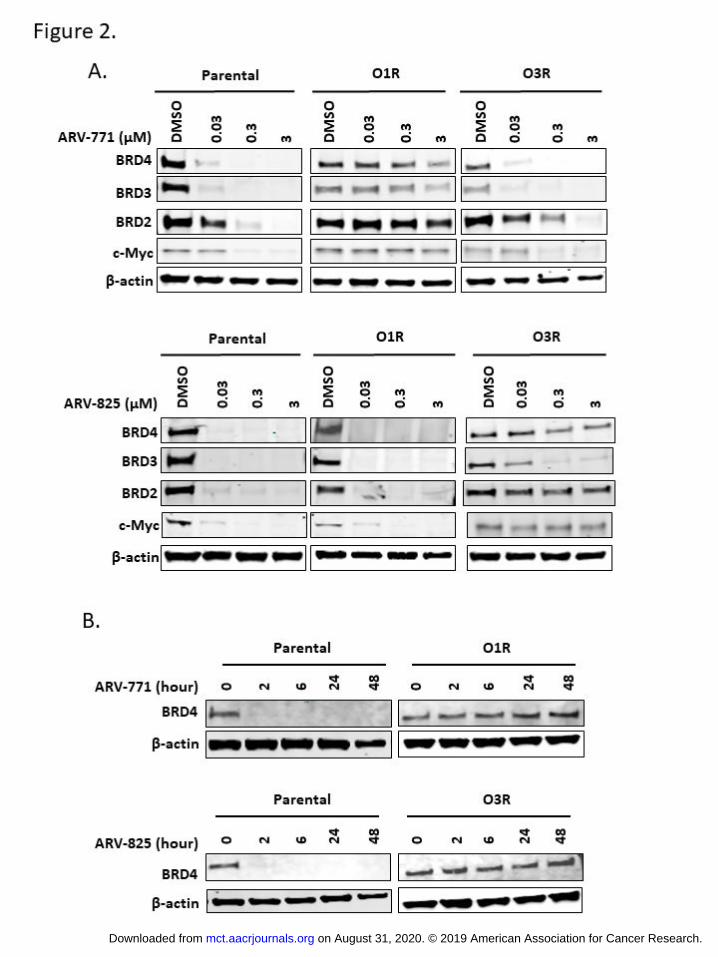
BET-PROTACs fail to induce BET protein degradation in the resistant cells. It has been shown that BET-
PROTACs inhibit cell growth and cause apoptosis by inducing degradation of BET family proteins. As
expected, treatment of ARV-771 for 16 hours caused degradation of BRD2, BRD3, BRD4, and inhibited
the well-established BET target gene Myc in the parental and the O3R cells in a dose-dependent manner
(Fig. 2A). In contrast, ARV-771 failed to induce significant BET protein degradation or Myc inhibition in
the O1R cells even at the very high concentration of 3 uM. Similarly, treatment of ARV-825 for 16 hours
caused BET protein degradation and Myc inhibition in the parental and O1R cells at as low as 30 nM (Fig.
2A). However, ARV-825 did not degrade BRD2 or BRD4 protein efficiently at up to 3 uM and only
partially degraded BRD3 protein in O3R cells at higher concentration (Fig. 2A). This is consistent with
previous reports in other cancer types that BRD3 is more readily degraded by BET-PROTACs compared
to BRD2 and BRD4 (6,24,26). Time course studies further established that extending BET-PROTAC
treatment to 48 hours still could not trigger BRD4 degradation in the resistant cells (Fig. 2B). These
results collectively demonstrate that the O1R and O3R cells are resistant to BET protein degradation
induced by VHL- or CRBN-based BET-PROTACs, respectively. The absence of cross resistance to both
VHL- and CRBN-based BET-PROTAC suggests that the proteasome degradation machinery downstream
of protein ubiquitination remains functional in these resistant cells.
Resistance to VHL-based BET-PROTAC is caused by Cullin 2(CUL2) loss due to multiple genomic
alterations at the CUL2 locus. To unravel the mechanism of resistance to VHL-based BET-PROTAC in the
O1R cells, we examined the genomic and transcriptional differences such as acquired mutations, gene
expression changes, and copy number variation (CNV) between the parental and O1R cells using whole
exome sequencing and RNA-seq. No genomic or mRNA expression alterations of BET family proteins or
VHL were found in the O1R cells, and the protein level of VHL is comparable in the parental and the O1R
cells (Supplementary Fig. S1D). Although the protein levels of BRD4 and BRD3 are slightly decreased in
O1R and O3R cells, there is still significant amount of these two proteins, especially BRD4, detected
(Supplementary Fig. S1D). Interestingly, the O1R cells were found to possess multiple genomic
alterations that impact the gene encoding cullin2, a critical component of the VHL CRL complex (Fig. 3A).
The alterations include a frame-shift mutation predicted to cause nonsense-mediated mRNA decay by
introducing a premature stop codon at position 442 in the cullin homology domain; an intronic mutation
upstream of exon 12 with concomitant skipping of exon 12; and a large-scale deletion of 42Mb
encompassing the CUL2 gene (Fig.3B-D). To investigate the copy number of this region, we used SNP
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

7
minor allele frequencies across chromosome 10 in the parental, O1R, and O3R cells (Supplementary Fig.
S3A). The pattern suggested the presence of other chromosomal aberrations preexisting in the parental,
making the estimation of exact copy number challenging. Follow-up experiments confirmed the frame
shift and the intronic mutations by Sanger sequencing and the copy number loss by TaqMan CNV
analysis (Fig. 3E; Supplementary Fig. S3C and D). Consistent with the abnormalities at the genomic level,
RNA-seq revealed CUL2 to be one of the most significantly down-regulated genes in the O1R cells
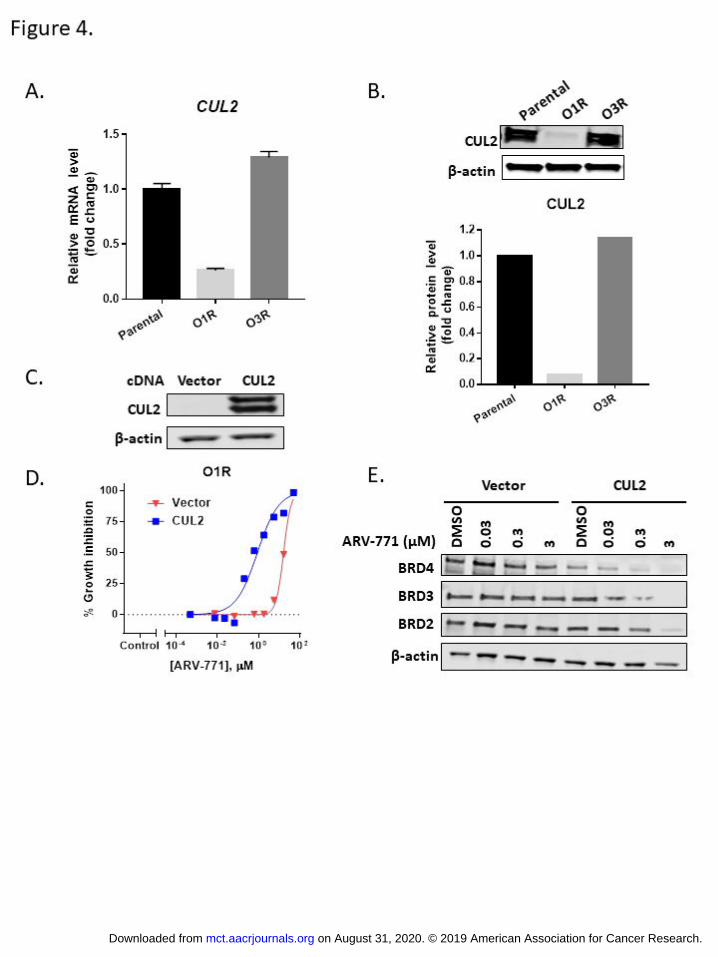
compared to parental cells (Supplementary Fig. S4A). Quantitative PCR and Western blot analysis further
established the significant reduction of CUL2 mRNA and CUL2 protein in the O1R cells compared to the
parental cells (Fig. 4A and B). In contrast, the O3R cells (resistant to the CRBN-based BET-PROTAC) were
devoid of these genomic abnormalities in the CUL2 gene, and expression of CUL2 was comparable to the
parental cells (Fig. 4A and B).
The hypoxia inducible factors HIF-1A and HIF-2A are physiological substrates of the VHL CRL
complex. Under normoxic conditions, the VHL CRL complex mediates HIF-1 ubiquitination and
degradation, consequently preventing HIF-1 dependent transcription under normoxia (27). GSEA
analyses revealed significant up-regulation and enrichment of the HIF1A and HIF2A pathways under
normoxic conditions in the O1R cells compared to the parental cells (Supplementary Fig. S4C). qPCR
analysis also demonstrated the up-regulation of HIF-1 target genes SOD2, VEGF, and GLUT1 in the O1R
cells compared to the parental cells or the O3R cells (Supplementary Fig. S4D). These results collectively
support that CUL2 loss in the O1R cells compromises the function of the VHL CRL complex.
To determine whether CUL2 loss directly contributes to the resistance to VHL-based BET-
PROTAC, we created O1R derived cell lines that express exogenous CUL2 (Fig. 4C). As shown in Fig. 4D
and E, overexpression of CUL2 in the O1R cells re-sensitized the cells to ARV-771 in the cell proliferation
assay and restored ARV-771-induced BET protein degradation, demonstrating that CUL2 loss is
responsible for acquired resistance in O1R cells.
Resistance to CRBN-based BET PROTAC is caused by the loss of Cereblon (CRBN) gene due to
chromosomal deletion. Whole exome sequencing and RNA-seq were also carried out to determine the
genomic and transcriptional differences such as acquired mutations, gene expression changes, and copy
number variation (CNV) between the O3R cells and the parental or the O1R cells. While no genomic or
expression alterations of BET family proteins were found in the O3R cells, a 12 Mb deletion on
chromosome 3 that encompasses the CRBN gene was found in O3R but not in the O1R cells (Fig.5A).
Based on SNP minor allele frequencies, copy number was estimated as 3 copies for chromosome 3p in
the parental and the O1R cells and 1 copy in the region containing CRBN of the O3R cells
(Supplementary Fig. S3B). The estimated loss of 2 copies was consistent with TaqMan CNV analysis,
which demonstrated a greater than 50% reduction in genomic DNA level relative to parental (Fig. 5B).
Consistent with the CRBN gene deletion, RNA-seq differential expression analysis identified CRBN as one
of the most significantly down-regulated genes in the O3R cells compared to parental cells
(Supplementary Fig. S4B). qPCR and Western blot analysis confirmed significant down-regulation of the
CRBN mRNA and CRBN protein in the O3R cells (Fig. 5C and D).
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

8
To determine whether the reduction of CRBN directly contributes to the resistance to CRBN-
based BET-PROTAC in the O3R cells, we created O3R derived cell lines that express high levels of CRBN
(Fig. 5E). As shown in Fig. 5F and G, increasing CRBN expression in the O3R cells re-sensitized the cells to
ARV-825 in proliferation assays and restored ARV-825 induced BET protein degradation.
Discussion
PROTACs have emerged as a promising novel modality for the development of next generation
therapeutics. In this study, we reported that resistance to both VHL- and CRBN-based BET-PROTACs can
occur in cancer cells following chronic treatment. However, unlike what is often observed for other
targeted therapeutics, such as kinase inhibitors, cells that were resistant to ARV-771 and ARV-825 did
not contain secondary mutations that affect compound binding to the target. Although the protein
levels of BRD3 and BRD4 are slightly decreased in O1R and O3R cells, there is still significant amount of
these two proteins, especially BRD4, detected. Considering that BRD4 is the major player in c-Myc
regulation among the three BRD proteins, this subtle change in BRD4 protein level is not likely to
contribute to the acquired drug resistance (28). Rather, resistance to both classes of BET-PROTACs was
primarily attributed to genomic alterations that impact core components of the corresponding E3 ligase
complexes. In cells that were resistant to ARV-771 or ARV-825, the proteasome degradation machinery
also remained intact. The preference of targeting the E3 ligases components over the proteasome
machinery for resistance development is intriguing. We suspect that the particular vulnerability of E3
ligase components for resistance development may relate to the redundancy and/or non-essentiality of
these components for cell survival, while compromising proteasome function in cells could be lethal or
significantly compromise cell fitness. The multiple genomic alterations at the CUL2 locus suggest the
existence of strong selection pressure for the cells to abolish CUL2 function in the VHL CRL complex for
resistance development. Interestingly, in cells that are resistant to the CRBN-based BET-PROTAC ARV-
825, resistance was primarily attributed to CRBN deletion and no genomic/transcription alterations were
observed for CUL4, the CUL2 equivalent of the CRBN CRL complex. CRBN appears to be a common point
of resistance development for CRBN-targeting agents. It has been shown that deletion of CRBN was the
primary cause of resistance to iMiDs in myeloma cells (29). The expression level of CRBN has also been
identified as a prognostic or predictive biomarker for multiple myeloma (MM) patients receiving iMiDs
(30-32). In addition to MM, both genetic alterations and differential expression of CUL2 and CRBN have
been observed in many other cancer types based on TCGA dataset (Supplementary Fig. S5) (33,34).
Interestingly, cell lines with lower CUL2 (Toledo and U266B1) or CRBN (TOV112D and HCT116)
expression are less sensitive to ARV-771 and ARV-825, respectively (Supplementary Fig. S6A and B).
Importantly, small interfering RNA (siRNA)-mediated knockdown of CUL2 or CRBN conferred resistance
to ARV-771 and ARV-825 in LNCaP cells, suggesting that there is an association between the expression
level of CUL2 or CRBN and sensitivity to VHL- or CRBN-based PROTACs (Supplementary Fig. S6C and D).
However, the expression level of these two proteins may not be the only determining factor for PROTAC
sensitivity. The fact that only the OVCAR8 cell line readily developed resistance to these compounds
may be attributed to the genetic background of the parental cells. Consistently, OVCAR8 cells harbor an
ATM mutation (p.V613L) and exhibit a mutational signature associated with double stranded break
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

9
(DSB) repair defects with elevated numbers of larger indels (>3bp) (Supplementary Fig. S7). These
underlying genetic attributes may play a role in the differential ability of the OVCAR8 cells to gain
resistance to the BET-PROTACs compared to other cell lines tested in this study.
In summary, we report here, for the first time, the mechanisms of acquired resistance to
PROTACs in cancer cell lines. These results highlight the critical involvement of E3 ligase complexes in
resistance development and lay the foundation for future investigation.
Acknowledgements:
We would like to thank Erin Murphy, Areej Ammar, Elina Dilmukhametova, and Ken Idler from the
AbbVie Genomic Technologies team for help in preparing the samples for sequencing analysis. Thank
you to Arne Grundstad from the AbbVie Computational Genomics team for help in processing the
exome sequencing data. A special thank you to Relja Popovic from the AbbVie Pharmacogenomics team
for key discussions guiding the initiation and design of the project.
References
1. Bosu DR, Kipreos ET. Cullin-RING ubiquitin ligases: global regulation and activation cycles. Cell division 2008;3:7 doi 10.1186/1747-1028-3-7.
2. Raina K, Crews CM. Chemical inducers of targeted protein degradation. The Journal of biological chemistry 2010;285(15):11057-60 doi 10.1074/jbc.R109.078105.
3. Fischer ES, Bohm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014;512(7512):49-53 doi 10.1038/nature13527.
4. Cardote TAF, Gadd MS, Ciulli A. Crystal Structure of the Cul2-Rbx1-EloBC-VHL Ubiquitin Ligase Complex. Structure 2017;25(6):901-11 e3 doi 10.1016/j.str.2017.04.009.
5. Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chemistry & biology 2015;22(6):755-63 doi 10.1016/j.chembiol.2015.05.009.
6. Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proceedings of the National Academy of Sciences of the United States of America 2016;113(26):7124-9 doi 10.1073/pnas.1521738113.
7. Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, et al. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. Journal of medicinal chemistry 2018;61(2):462-81 doi 10.1021/acs.jmedchem.6b01816.
8. Sun Y, Zhao X, Ding N, Gao H, Wu Y, Yang Y, et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell research 2018;28(7):779-81 doi 10.1038/s41422-018-0055-1.
9. Zhang C, Han XR, Yang X, Jiang B, Liu J, Xiong Y, et al. Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). European journal of medicinal chemistry 2018;151:304-14 doi 10.1016/j.ejmech.2018.03.071.
10. Sun B, Fiskus W, Qian Y, Rajapakshe K, Raina K, Coleman KG, et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 2018;32(2):343-52 doi 10.1038/leu.2017.207.
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

10
11. Saenz DT, Fiskus W, Qian Y, Manshouri T, Rajapakshe K, Raina K, et al. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia 2017;31(9):1951-61 doi 10.1038/leu.2016.393.
12. Churcher I. Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones? Journal of medicinal chemistry 2018;61(2):444-52 doi 10.1021/acs.jmedchem.7b01272.
13. Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nature chemical biology 2015;11(8):611-7 doi 10.1038/nchembio.1858.
14. Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015;348(6241):1376-81 doi 10.1126/science.aab1433.
15. Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, et al. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell chemical biology 2018;25(1):78-87 e5 doi 10.1016/j.chembiol.2017.09.010.
16. McDaniel KF, Wang L, Soltwedel T, Fidanze SD, Hasvold LA, Liu D, et al. Discovery of N-(4-(2,4-Difluorophenoxy)-3-(6-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin -4-yl)phenyl)ethanesulfonamide (ABBV-075/Mivebresib), a Potent and Orally Available Bromodomain and Extraterminal Domain (BET) Family Bromodomain Inhibitor. Journal of medicinal chemistry 2017;60(20):8369-84 doi 10.1021/acs.jmedchem.7b00746.
17. Ewels P, Magnusson M, Lundin S, Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016;32(19):3047-8 doi 10.1093/bioinformatics/btw354.
18. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome research 2012;22(3):568-76 doi 10.1101/gr.129684.111.
19. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15-21 doi 10.1093/bioinformatics/bts635.
20. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30(7):923-30 doi 10.1093/bioinformatics/btt656.
21. Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome biology 2014;15(2):R29 doi 10.1186/gb-2014-15-2-r29.
22. Su S, Law CW, Ah-Cann C, Asselin-Labat ML, Blewitt ME, Ritchie ME. Glimma: interactive graphics for gene expression analysis. Bioinformatics 2017;33(13):2050-2 doi 10.1093/bioinformatics/btx094.
23. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nature biotechnology 2011;29(1):24-6 doi 10.1038/nbt.1754.
24. Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014;510(7504):278-82 doi 10.1038/nature13229.
25. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011;478(7370):524-8 doi 10.1038/nature10334.
26. Bai L, Zhou B, Yang CY, Ji J, McEachern D, Przybranowski S, et al. Targeted Degradation of BET Proteins in Triple-Negative Breast Cancer. Cancer research 2017;77(9):2476-87 doi 10.1158/0008-5472.CAN-16-2622.
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

11
27. Kapitsinou PP, Haase VH. The VHL tumor suppressor and HIF: insights from genetic studies in mice. Cell death and differentiation 2008;15(4):650-9 doi 10.1038/sj.cdd.4402313.
28. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011;146(6):904-17 doi 10.1016/j.cell.2011.08.017.
29. Zhu YX, Braggio E, Shi CX, Bruins LA, Schmidt JE, Van Wier S, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011;118(18):4771-9 doi 10.1182/blood-2011-05-356063.
30. Broyl A, Kuiper R, van Duin M, van der Holt B, el Jarari L, Bertsch U, et al. High cereblon expression is associated with better survival in patients with newly diagnosed multiple myeloma treated with thalidomide maintenance. Blood 2013;121(4):624-7 doi 10.1182/blood-2012-06-438101.
31. Bila J, Sretenovic A, Jelicic J, Tosic N, Glumac I, Fekete MD, et al. Prognostic Significance of Cereblon Expression in Patients With Multiple Myeloma. Clinical lymphoma, myeloma & leukemia 2016;16(11):610-5 doi 10.1016/j.clml.2016.08.007.
32. Heintel D, Rocci A, Ludwig H, Bolomsky A, Caltagirone S, Schreder M, et al. High expression of cereblon (CRBN) is associated with improved clinical response in patients with multiple myeloma treated with lenalidomide and dexamethasone. British journal of haematology 2013;161(5):695-700 doi 10.1111/bjh.12338.
33. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 2013;6(269):pl1 doi 10.1126/scisignal.2004088.
34. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2(5):401-4 doi 10.1158/2159-8290.CD-12-0095.
35. Chen Z, Sui J, Zhang F, Zhang C. Cullin family proteins and tumorigenesis: genetic association and molecular mechanisms. Journal of Cancer 2015;6(3):233-42 doi 10.7150/jca.11076.
Figure legends
Figure 1. Cancer cells develop drug resistance after chronic exposure to BET-PROTACs. A. Sensitivity to
ARV-771 or ARV-825 of parental OVCAR8 (black), resistant derivatives O1R (blue) and O3R (red) was
assessed by CTG assays. Data represents mean±standard deviation (SD). N=3. The table lists the IC50
value fold change of resistant cells over parental cells for each compound. B. Cells were treated with
indicated concentration of ARV-771 or ARV-825 for 48 hours and harvested for Western blot analysis
using cleaved PARP antibody. β-actin was used as loading control. C. Caspase 3/7 assay was performed
in parental and resistant cells treated with a series dilution of ARV-771 or ARV-825 for 24 hours. Data
represents mean±standard deviation (SD). N=3.
Figure 2. BET-PROTACs fail to induce BET protein degradation in the resistant cells. A. Cells were
treated with indicated concentration of ARV-771 or ARV-825 for 16 hours and harvested for Western
blot analysis using antibodies against BET family proteins and c-Myc. β-actin was used as loading
control. B. Cells were treated with 0.05µM of ARV-771 or ARV-825 for indicated time and harvested for
Western blot analysis using antibodies against BRD4. β-actin was used as loading control.
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

12
Figure 3. Exome sequencing identified mutations and deletion in CUL2 gene. A. A model of CRL2 ligase.
Adapted from Chen. Et al. (35) B. CUL2 somatic mutations identified through exome sequencing of
parental and O1R samples. C. Sashimi plot of RNA-seq read coverage of CUL2 exons 11-14. Exon 12
skipping present in O1R replicates (red) but not in Parental replicates (black). Solid peaks depict reads
per kilobase per million reads mapped (RPKM) within individual exons. Reads that bridge different exons
are shown as arcs. Intron deletion and T435Hfs deletion indicated by blue and red arrows, respectively.
D. CUL2 deletion detected in O1R by copy number calling on exome sequencing data (relative to
parental). E. CUL2 deletion confirmation by TaqMan CNV assay.
Figure 4. Resistance towards VHL based BET PROTAC ARV-771 derives from depletion of CUL2. A. qPCR
analysis of CUL2 mRNA level in parental, O1R and O3R cells. Data represents mean±standard deviation
(SD). N=3. B. Western blot analysis of CUL2 protein level in parental, O1R and O3R cells. β-actin was
used as loading control. Quantification of CUL2 protein level is shown below. C. Western blot analysis of
CUL2 in O1R cells expressing empty vector or CUL2 cDNA. β-actin was used as loading control. D. Dose
response curves for ARV-771 in O1R cells expressing empty vector or CUL2 cDNA. Data represents
mean±standard deviation (SD). N=3. E.O1R cells expressing empty vector or CUL2 cDNA were treated
with indicated concentration of ARV-771 for 16 hours and harvested for Western blot analysis. β-actin
was used as loading control.
Figure 5. Resistance to CRBN-based BET PROTAC is caused by the loss of Cereblon (CRBN) gene due to
chromosomal deletion. A. CNV plot of chromosome 3 from O3R cells. B. CRBN deletion confirmation by
TaqMan CNV assay. C. qPCR analysis of CRBN mRNA level in parental, O1R and O3R cells. Data
represents mean±standard deviation (SD). N=3. D. Western blot analysis of CRBN level in parental, O1R
and O3R cells. β-actin was used as loading control. Quantification of CRBN protein level is shown below.
E. Western blot analysis of CRBN in O3R cells expressing empty vector or CRBN cDNA. β-actin was used
as loading control. F. Dose response curves for ARV-825 in O3R cells expressing empty vector or CRBN
cDNA. Data represents mean±standard deviation (SD). N=3. G.O3R cells expressing empty vector or
CRBN cDNA were treated with indicated concentration of ARV-825 for16 hours and harvested for
Western blot analysis. β-actin was used as loading control.
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129

Published OnlineFirst May 7, 2019.Mol Cancer Ther Lu Zhang, Bridget Riley-Gillis, Priyanka Vijay, et al. of E3 ligase Complexes
ComponentsChimeras) Caused by Genomic Alterations in Core Acquired Resistance to BET-PROTACs(Proteolysis Targeting
Updated version
10.1158/1535-7163.MCT-18-1129doi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2019/05/07/1535-7163.MCT-18-1129.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/early/2019/05/07/1535-7163.MCT-18-1129To request permission to re-use all or part of this article, use this link
on August 31, 2020. © 2019 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on May 7, 2019; DOI: 10.1158/1535-7163.MCT-18-1129