ab initio based modeling of advanced materials amm …iqms.ru/registration/programme.pdf · ab...
TRANSCRIPT

AB INITIO BASED MODELING OF
ADVANCED MATERIALS
AMM-2016
Satellite Conference of XX Mendeleev Congress on general
and applied chemistry
Ekaterinburg
22 – 24 September, 2016

–2–
Organizers
Institute of Quantum Institute of Metal Physics
Materials Science CJSC Ural Branch RAS
Ural Hi-Tech Park Ural Federal University
Chairmans
Yuri Gornostyrev (Ekaterinburg, Russia)
Mikhail Katsnelson (Nijmegen, Netherlands)
The conference brings together prominent scientists from the area of theoretical modelling to
assess the state of the art in applications of the electronic structure theory for the knowledge-
based design of advanced materials. Particularly the conference will focus on the development
of theoretical approaches and physical principles of accelerated materials design, discussion of
novel trends in materials science and promote direct interaction between theory and experiment.
The aim is to provide a distinguished atmosphere and framework for exchanging the newest
ideas and concepts in order to resolve the present challenges in the field.
Master class by Alexander I. Poteryaev the “Application of the AMULET code for DFT+DMFT
calculations of realistic compounds” will be held during the conference.
This conference is supported and financed by
ART, SCIENCE AND SPORT CHARITY
FOUNDATION
SVERDLOVSK REGION ADMINISTRATION
RTC AUSFERR
ISBN - 978-5-7691-2455-6
URAL BRANCH RAS

–3–
AMM-2016 conference program
22 September
9:30 –10:30 Registration. Coffee is served
10:30 –10:45 Opening. Welcome talk
SESSION 1: DMFT AND ALL THAT
10:45–12:40 Chair Mikhail Katsnelson
10:45 –11:25 Alexander Lichtenstein, University Hamburg, Hamburg, Germany
Strong Electronic Correlations in magnetic materials
11:25 –11:50 Andrey Katanin, IMP Ural Branch RAS, Ural Federal University,
Ekaterinburg, Russia
Non-local effects in strongly-correlated systems
11:50 –12:15 Ivan Leonov, University of Augsburg, Augsburg, Germany
Electronic structure and phase stability of correlated electron materials under
extreme conditions
12:15 –12:40 Sergey Streltsov, Igor Mazin, IMP Ural Branch RAS, Russia, NRL, USA
Molecular orbitals in hexagonal ruthenates
13:00 –14:00 Lunch
14:00-16:10 Chair Tamio Oguchi
14:00 –14:40 Vladimir Anisimov, IMP Ural Branch RAS, Ekaterinburg, Russia
Magnetic and structural properties of iron and its alloys in LDA+DMFT
14:40 –15:20 Olle Eriksson, Uppsala university, Uppsala, Sweden
Theory of x-ray and photoelectron spectroscopy with DMFT
15:20 –15:45 Leonid Pourovskii, CPHT-Ecole Polytechnique, CNRS, Université Paris-
Saclay, France
Impact of electronic correlations on point-defect thermodynamics and
transport in iron metal

–4–
15:45 –16:10 Igor Nekrasov, Institute of Electrophysics Ural Branch RAS, Ekaterinburg
Magnetocaloric effect in strongly correlated systems
16:10 –16:40 Coffee break
16:40-17:55 Chair Vladimir Anisimov
16:40 –17:05 Alessandro Toschi, Institute of Solid State Physics, TU Wien, Austria,
Quantum many-body theory at the two-particle level
17:05 –17:30 Georg Rohringer, Vienna University of Technology, Wien, Austria
Impact of non-local correlations over different energy scales: a dynamical
vertex approximation study
17:30 –17:55 Alexey Rubtsov, Russia Quantum Center, Moscow, Russia
Modeling of the impurity dynamics in ultracold atomic media
18:00 –20:00 Poster session and reception (beer and food)
23 September
SESSION 2: MAGNETISM AND ALL THAT
10:00-12:20 Chair Alexander Lichtenstein
10:00 –10:40 Tamio Oguchi, ISIR, Osaka university, Japan
Electronic properties associated with spin-orbit coupling and broken symmetry
10:40 –11:05 Igor Solovyev, National Institute for Materials Science, Tsukuba, Japan
Origin and microscopic mechanisms of magnetoelectric coupling in
multiferroic manganites
11:05 –11:30 Vladimir Mazurenko, Ural Federal University, Ekaterinburg, Russia
Methods for calculation and analysis of the Dzyaloshinskii-Moriya interaction

–5–
11:30 –11:55 Mikhail Korotin, Institute of Metal Physics, Ekaterinburg, Russia
Coherent potential approximation for strongly correlated systems with spin-
orbit coupling
11:55 –12:35 Sergey Savrasov, University of California, Davis, CA, USA
Weyl semi-metal: a new topological state in condensed matter.
13:00 –14:00 Lunch
14:00-16:05 Chair Olle Eriksson
14:00 –14:25 Alexander Tsirlin, University of Augsburg, Augsburg, Germany
Ab initio evaluation and experimental verification of magnetic exchange
parameters in insulators
14:25 – 14:50 Sergey Skornyakov, Institute of Metal Physics, Ekaterinburg, Russia
Electronic correlations and topological Fermi surface transition in the iron-
based chalcogenides
14:50 – 15:15 Sergey Khmelevskyi, Vienna University of Technology, Wien, Austria
Functional antiferromagnetic materials for spintronics applications: challenge
for ab initio computations.
15:15 – 15:40 Tilmann Hickel, X. Zhang, J. Rogal, Jörg Neugebauer, MPIE, Dusseldorf,
Germany
The role of interfaces for structural transformations among austenite, ferrite
and cementite in Fe-C alloys
15:40 –16:10 Coffee break
16:10 –18:40 Master class
Alexander Poteryaev, IMP Ural Branch RAS, Ekaterinburg, Russia
Application of the AMULET code for DFT+DMFT calculations of realistic
compounds

–6–
24 September
SESSION 3: ALLOYS, STRUCTURE AND ALL THAT
10:00-12:25 Chair Pavel Korzhaviy
10:00 –10:40 Jörg Neugebauer, A. Glensk, F. Koermann, B. Grabowski, T. Hickel, MPIE,
Dusseldorf, Germany
Ab initio thermodynamic description of advanced structural materials: Status
and challenges
10:40 –11:20 Igor Abrikosov, Linköping University, Linköping, Sweden
Finite temperature effects in ab initio simulations of alloy thermodynamics
11:20 –11:45 Alexander Rudenko, Radboud University, Nijmegen, Netherlands
Intrinsic transport properties of monolayer black phosphorus
11:45 –12:10 Danil Boukhvalov, Department of Chemistry, Hanyang University, Korea
Locally destroyed crystal order in Ti-Fe alloys.
12:10 –12:35 Alexander Mirzoev, A. Verkhovykh, South Ural State University,
Chelyabinsk, Russia
The interaction of hydrogen interstitials with grain boundaries in bcc iron
13:00 –14:00 Lunch
14:00-16:05 Chair Igor Abrikosov
14:00 –14:25 Sergey Simak, Linköping University, Linköping, Sweden
Temperature-driven martensitic phase transitions from first principles
14:25 –14:50 Vsevolod Razumovskiy, D. Scheiber, L. Romaner, Materials Center Leoben
Forschung GmbH (MCL), Leoben, Austria
Impurity segregation and its effect on the grain boundary embrittlement in Ti:
effects of chemical and structural contributions
14:50 –15:15 Nadezhda Medvedeva, Institute of Solid State Chemistry, Ekaterinburg, Russia
Ab initio simulation of phosphorus in bulks, at surfaces and interface of fcc Fe
and K-carbide
15:15 –15:40 Oleg Gorbatov, Institute of Quantum Materials Science, Ekaterinburg, Russia
Effect of composition on antiphase boundary energy in NI3AL based alloys
15:40 –16:05 Mikhail Petrik, Institute of Metal Physics Ural Branch RAS; Institute of
quantum materials science, Ekaterinburg, Russia

–7–
Ab initio investigation of grain boundary segregation in Al=X
(X=Mg,Zn,Si,Cu) alloys
16:05 –16:30 Coffee break
16:30-18:40 Chair Yuri Gornostyrev
16:30 –17:10 James Morris, Oak Ridge National Lab, Oak Ridge, TN, USA
Ab initio modeling for understanding and predicting novel alloy behavior
17:10 –17:50 Pavel Korzhavyi, Royal Institute of Technology, Stockholm, Sweden
Ab initio based models of disordered materials
17:50 –18:15 Mikheil Sekania, University of Augsburg, Augsburg, Germany
Scaling behavior of the Compton profile of alkali metal elements
Poster Session and Reception
22 September, 18:00 – 20:00
1 V. Greshnyakov, E. Belenkov, Chelyabinsk State University, Chelyabinsk, Russia
Ab initio modelling of diamond-like materials
2 M. Shundalov, A. Matsukovich, S. Gaponenko, Belarusian State University; B.I.
Stepanov Institute of Physics, Minsk, Belarus
DFT and multi-reference perturbation theory calculations of the structures and uv-vis
spectra of adamantane-containing molecules, potential antibacterial agents
3 A. Gerasimov, V. Mazurenko, S. Skornyakov, Ural Federal University; Institute of Metal
Physics Ural Branch RAS, Ekaterinburg, Russia
Modelling of the magnetic interaction of strongly correlated systems
4 I. Kashin, I. Solovyev, V. Mazurenko, Ural Federal University, Ekaterinburg, Russia;
National Institute for Materials Science, Tsukuba, Japan

–8–
Effect of dynamical electron correlations on collective magnetic excitations in CrO2
5 S. Andreev, I. Solovyev, V. Mazurenko, Ural Federal University, Ekaterinburg, Russia;
National Institute for Materials Science, Tsukuba, Japan
Pressure dependence of the electronic structures of Sr3Ir2O7
6
O. Sotnikov, V. Mazurenko, Ural Federal University, Ekaterinburg, Russia
A method for calculating paramagnetic exchange interactions
7 I. Nekrasov, N. Pavlov, M. Sadovskii, A. Slobodchikov, Institute of Electrophysics of the
Ural Branch RAS; Institute of Metal Physics Ural Branch RAS, Ekaterinburg, Russia
The electronic structure of a monolayer FeSe on the SrTiO3 substrate
8 V.V. Bannikov, V.S. Kudyakova, A.A. Elagin, M.V. Baranov, A.R. Beketov , Ural
Federal University, Ekaterinburg, Russia; University of Michigan, USA; Hamburg
University, Germany
Electronic structure and magnetic properties of hexagonal and cubic modifications of
aluminium nitride doped with sp-impurities (B, C, O)
9 D. Medvedeva, V. Mazurenko, S. Iskakov, A. Lichtenstein, Ural Federal University,
Ekaterinburg, Russia; University of Michigan, USA; Hamburg University, Germany
Calculation scheme based on the extended equations of DMFT for square and triangular
lattices
10 J. Komleva, S. Nikolaev, A. Tsirlin, V. Mazurenko, Ural Federal University,
Ekaterinburg, Russia; University of Augsburg, Germany
Lattice dynamics in copper chloride CuCl2
11 D. Badrtdinov, S. Nikolaev, V. Mazurenko, Ural Federal University,
Ekaterinburg, Russia
Spin-orbit coupling effects in adatom systems Si(111):{C,Si,Sn,Pb}
12 D. Zakir’yanov, V. Chernyshev, Ural Federal University, Russia
Lead oxyhalides Pb3X2O2 (X=Cl, Br, I): ab initio calculations of phonon spectra and
optical properties
13 A. Stepanenko, D. Vesnina, P. Igoshev, A. Katanin, Ural Federal University, Institute of
Metal Physics Ural Branch RAS, Ekaterinburg, Russia
The Kohn anomalies in three-dimensional systems
14 V. Protsenko, A. Katanin, Institute of Metal Physics Ural Branch RAS; Ural Federal
University, Ekaterinburg, Russia
Electron transport through double quantum dots: the functional renormalization group
approach

–9–
15 D. Prishchenko, A. Rudenko, V. Mazurenko, M. Katsnelson, Ural Federal University,
Ekaterinburg, Russia; Radboud University, Nijmegen, Netherlands
Plasmons and screening in phosphorus: beyond wavelength limit
16 S. Sozykin, V. Beskachko, South Ural State University, Chelyabinsk, Russia
Contacts of carbon and gold nanotubes: first principles calculations
17 I. Tikina, N. Barbin, Ural Institute of state fire service of EMERCOM of Russia; Ural
state agrarian University, Ekaterinburg, Russia
Thermodynamic modeling of thermal dissociation of the intermetallic compounds
PbBi2Sn2
18 M. Petrik, D. Badrtdinov, Institute of Metal Physics Ural Branch RAS; Institute of
quantum materials science; Ural Federal University, Ekaterinburg, Russia
Magnetic anisotropy effects in Fe-Ga alloys
19 D. Nazipov, A. Nikiforov, L. Gonchar, Ural Federal University; Ural State University of
Railway Transport, Ekaterinburg, Russia
Structure and lattice dynamics of BiMnO3: Ab initio calculations
20 V. Chernyshev, A. Nikiforov, V. Petrov, Ural Federal University, Ekaterinburg, Russia
Structure and lattice dynamics of PrFe3(BO3)4: Ab initio calculation
21 I. Leonidov, V. Petrov, V. Chernyshev, A. Ishchenko, E. Konstantinova, A. Nikiforov,
Institute of Solid State Chemistry UB RAS; Ural Federal University, Ekaterinburg,
Russia
Lanthanide-Doped Germanates: DFT Study of Lattice Dynamics and Electronic
Structure of Optical Hosts
22 Z. Pchelkina, O. Volkova, V. Mazurenko, A. Vasiliev, Institute of Metal Physics Ural
Branch RAS, Ural Federal University, Ekaterinburg, Lomonosov Moscow State
University, National University of Science and Technology “MISiS,” Moscow, Russia
Electronic structure and magnetic properties of the strong-rung spin-1 ladder
Rb3Ni2(NO3)7
23 I. Piterskikh, D. Boukhvalov, V. Mazurenko, Ural Federal University, Ekaterinburg,
Russia; Hanyang University, Seoul, Republic of Korea
Full potential study of electronic and magnetic properties of functionalized graphene
24 D. Suetin, Institute of Solid State Chemistry, Ural Branch RAS, Ekaterinburg, Russia
Structural, electronic properties, stability and fermi surfaces of ternary borides CaM2B2,
CaM3B2, Ca2M5B4, Ca3M8B6 (M = Rh, Ir)
25 M. Ivonina, P. Snegurov, V. Sizov, Saint Petersburg State University, Saint Petersburg,
Russia
Oxygen ion diffusivity in scandia-stabilized zirconia: molecular dynamics simulations

–10–
and ab initio calculation
26 L. Kar’kina, I. Kar’kin, A. Kuznetsov, Institute of Metal Physics Ural Branch RAS;
Institute of quantum materials science, Ekaterinburg, Russia
Atomistic simulation of stacking faults in cementite
27 A. Stroev, Yu. Gornostyrev, Institute of Metal Physics Ural Branch RAS; Institute of
quantum materials science, Ekaterinburg, Russia
Precipitation kinetics and GPZ formation in Al-based alloys. Master equation approach
with ab-initio parameterization
28 I. Shmakov, I. Razumov, Yu. Gornostyrev, Institute of Metal Physics Ural Branch RAS;
Institute of quantum materials science, Ekaterinburg, Russia
Decomposition kinetics in Fe–Cu dilute alloys. Monte Carlo simulations
29 K. Nekrasov, N. Kichigina, Ural Federal University, Ekaterinburg, Russia
Molecular dynamics simulation of bulk xenon diffusion in UO2: a comparison of ab initio
interaction potentials
30 I. Lomaev, D. Novikov, S. Okatov, Yu. Gornostyrev, S Burlatsky, Institute of Quantum
Materials Science, Institute of Metal Physics UB RAS, Ekaterinburg, Russia, United
Technologies Research Center (UTRC), USA
Size misfit versus electronic effects in diffusion of substitutional impurities in ni matrix
31 A. Kardashin, V. Mazurenko, Ural Federal University, Ekaterinburg, Russia
Electronic and magnetic properties of iron impurities on W(110)
32 A. Pravednicov, A. Tsirlin, D. Prishchenko, V.G. Mazurenko, Experimental Physics VI,
Center for Electronic Correlations and Magnetism, Institute of Physics, University of
Augsburg, Germany; Ural Federal University, Ekaterinburg, Russia
Modeling the electronic structure and dynamics of the crystal lattice TiPO₄

ABSTRACTS
INVITED AND CONTRIBUTED TALKS

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–12–
STRONG ELECTRONIC CORRELATIONS IN MAGNETIC MATERIALS
Alexander Lichtenstein,
University of Hamburg, Germany
Effects of electron interactions in magnetic materials, oxides and transition metals will
be discussed. Modern density functional theory describes well the ground state properties for
moderate correlated metals, but failed for some Mott insulators. Spectroscopy of strongly
correlated magnetic materials with transition or rare-earth elements can be well incorporated
only in correlated electronic structure scheme. We introduce a multi-orbital spin-polarized
dynamical mean field theory which allowed investigating the correlations effects in real
materials. Prospects of realistic description of itinerant magnetism in transition metals and Mott
insulators state in complex oxides will be discussed.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–13–
NON-LOCAL EFFECTS IN STRONGLY-CORRELATED SYSTEMS Andrey Katanin1,2
1Institute of Metal Physics, 620990, Ekaterinburg, Russia
2Ural Federal University, 620002, Ekaterinburg, Russia
We consider non-local effects in strongly-correlated systems and theoretical approaches,
which allow for their description based on dynamical mean-field theory as a starting point.
Although some of such approaches ((E)DMFT+GW, dynamic vertex approximation, dual
fermion/boson approaches) are known for relatively long time, we consider newer DMFT+spin-
fermion model, DMF2RG, and (E)DMFT+2PI fRG approaches.
In the first part of the talk we discuss effects of non-local magnetic correlations in iron. As a
theoretical tool, we consider DMFT+spin-fermion model approach [1,2]. This approach allows
evaluating non-local (Heisenberg-type) exchange interactions and Curie temperature, which
agree well with existing estimates, but in addition one can obtain information on the non-local
contributions to energy, free energy, etc. This approach is also applied to describe non-local
correlations in the vicinity of - structural transformation in iron.
In the second part of the talk we consider theoretical approaches to screening of Coulomb
interaction beyond (E)DMFT+GW. In particular, we describe the (E)DMFT+2PI fRG approach
[3], which treats the self-energy and polarization operator corrections to DMFT, using functional
renormalization-group method for the two-particle irreducible vertices. Being close in spirit to
dynamic vertex approximation [4] from one side (regarding using of the two-particle irreducible
vertices) and DMF2RG approach [5] from the other side (regarding using functional
renormalization group method), the described approach allows treatment of non-local
interactions in correlated electronic systems in a scheme, which accounts for vertex corrections
over the standard applications of (E)DMFT+GW approach. The resulting equations and the
relation to the dual boson approach are discussed.
[1] Igoshev PA, Efremov AV, Katanin AA, Phys. Rev. B 2015; 91:195123
[2] Katanin AA, Belozerov AS, Anisimov VI, arXiv:1605.04589.
[3] Katanin AA, ArXiv: 1604.01702.
[4] Toschi A, Katanin AA, Held K, Phys. Rev. B 2007; 75:045118;
Toschi A, Rohringer G, Katanin AA, Held K, Ann. der Phys. 2011; 523:698.
[5] Taranto C, Andergassen S, Bauer J, Held K, Katanin A, Metzner W, Rohringer G, Toschi
A, Phys. Rev. Lett. 2014; 112:196402.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–14–
ELECTRONIC STRUCTURE AND PHASE STABILITY OF
CORRELATED ELECTRON MATERIALS UNDER EXTREME
CONDITIONS Ivan Leonov 1,2*
1 Theoretical Physics III, Center for Electronic Correlations and Magnetism, University of
Augsburg, Germany 2 Materials Modeling and Development Laboratory, National University of Science and
Technology 'MISIS', 119049 Moscow, Russia
Computational studies of electronic correlations and magnetism and, in particular, a realistic
modelling of strongly correlated electron materials, is a challenging theoretical problem. In this
talk, I will discuss an application of the novel computational scheme LDA+DMFT to explore the
electronic and structural properties of correlated materials [1]. In particular, I will present our
recent results for the pressure-induced magnetic collapse and Mott insulator-metal transition in
paramagnetic oxides MnO, FeO, CoO, and NiO, and the electronic structure and phase stability
of Fe2O3 near a pressure-induced Mott metal-insulator transition. Our results for the electronic
state, the equilibrium crystal structure, and the structural phase stability are in quantitative
agreement with experimental data. We find that electronic correlations are important to explain
the lattice stability of correlated materials.
[1] Leonov I, et al, Phys Rev Lett 2011; 106:106405; Leonov I, et al, Phys Rev Lett 2014;
112:146401; Leonov I, et al, Phys Rev Lett 2015; 115: 106402; Leonov I, et al, Phys Rev B
2015; 91:195115.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–15–
MOLECULAR ORBITALS IN HEXAGONAL RUTHENATES Sergey Streltsov 1*, Igor Mazin 2
1 Institute of Metal Physics Ural Branch RAS, Russia
2 Naval Research laboratory, USA
We show that unique magnetic properties properties of several layered hexagonal ruthenates
including Li2RuO3 and SrRu2O6 can be explained basing on the conception of the molecular
orbitals. In Li2RuO3 formation of the molecular orbitals results in dimerization, appearance of
the spin gap and strong decrease of the magnetic susceptibility. Surprisingly, molecular orbitals
survive even at much higher temperature, where valence bond liquid state stabilizes. In this state
thermal fluctuations drive resonance between different dimer coverages, a classic analog of the
resonating valence bond state often discussed in connection with high-Tc cuprates [1].
Very different situation is observed in SrRuO6, which attract a lot of attention due to
surprisingly large Neel temperature ~550K despite of the layered structure. First principles
calculations show that only an ideal Neel ordering in the Ru plane is possible, with no other
metastable magnetic solutions, and, highly unusually, yield dielectric gaps for both
antiferromagnetic and nonmagnetic states. We demonstrate that this strange behavior is the
result of the formation of very specific electronic objects, molecular orbitals, whereby each
electron is well localized on a particular Ru6 hexagon, and completely delocalized over the
corresponding six Ru sites, thus making the compound both strongly localized and highly
itinerant [2].
[1] Kimber S.A.J., Mazin I.I., Shen J., Jeschke H.O., Streltsov S.V., Argyriou D.N., Valenti R.,
Khomskii D.I., Phys. Rev. B 2014 89: 081408
[2] Streltsov S., Mazin I.I., Foyevtsova K. Phys. Rev. B 2015, 92:134408
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–16–
MAGNETIC AND STRUCTURAL PROPERTIES OF IRON AND ITS
ALLOYS IN LDA+DMFT Vladimir Anisimov
Institute of Metal Physics Ural Branch RAS, Ekaterinburg, Russia
Iron is a metal with a well-defined local moment and its magnetic properties above the Curie
temperature can be well described by LDA+DMFT method. We discuss the problem of
determining the values of direct and exchange Coulomb parameters U and J and dependence of
the calculation results on them. Correlation effects strongly influence not only magnetic but also
structural properties of iron. We present results of LDA+DMFT calculations for alpha-gamma
and gamma-delta transitions. Combination of Coherent Potential approach (CPA) with
LDA+DMFT allows treating correlation effects for alloys of iron. It was applied to the problem
of structural transition in iron-manganese alloy and also to calculation of Curie temperature for
iron-nickel alloy.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–17–
THEORY OF X-RAY AND PHOTOELECTRON SPECTROSCOPY WITH
DMFT Olle Eriksson
Department of Physics and Astronomy, Uppsala university, Uppsala, Sweden
In this talk I will present the basic ideas of dynamical mean-field theory (DMFT) and its
implementation in a full-potential electronic structure method that uses linear muffin-tin orbitals
as basis functions. Examples of DMFT calculations of the electronic structure calculated will be
given, for rare-earth as well as transition metal oxides. For several materials I will compare the
DMFT results to more traditional calculations like LSDA and LDA+U. The extension of this
theory to describe x-ray absorption processes will also be given with calculated examples from
the transition metal monoxides. The class of materials covered in this talk range from bulk to
clusters.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–18–
IMPACT OF ELECTRONIC CORRELATIONS ON POINT-DEFECT
THERMODYNAMICS AND TRANSPORT IN IRON METAL Leonid Pourovskii
CPHT-Ecole Polytechnique, CNRS, Université Paris-Saclay,
F-91128 Palaiseau, France
We apply an ab initio theoretical framework combining the density functional and dynamical
mean field theories (DFT+DMFT) to study point-defect thermodynamics and transport
properties of elemental iron.
First, we consider hexagonal close-packed ε-iron at the Earth's core volume and for
temperatures up to 7000K [1]. We find that highly compressed ε-Fe behaves as a nearly perfect
Fermi liquid with the corresponding Fermi-liquid temperature scale TFL of about 14000 K, i.e.
much higher than the possible temperature range for the core. The calculated electron-electron-
scattering contribution to the electrical resistivity is rather insignificant compared to the
electron-phonon one. However, we find that the electron-electrons-scattering still quite
important for the thermal resistivity of ε-Fe, which is greatly enhanced due to the Fermi-liquid
quadratic frequency dependence of the scattering rate. Hence, the Fermi liquid behavior causes
the calculated thermal resistivity to be of comparable magnitude to the electron-phonon one.
One may expect a significant impact due to this effect on the dynamics of Earth’s core.
Another application of the same approach concerns the single-vacancy formation energy in
α-Fe at ambient conditions. We find that the vacancy formation energy is substantially reduced
as compared to previous standard density-functional theory calculations, with the obtained
theoretical value being in excellent agreement with experiment. The reduction is found to be
induced by enhancement of correlations at the vacancy's nearest-neighbors; this enhancement is
explained by subtle changes in the corresponding local density of states of d-electrons. Local
lattice relaxations around the vacancy are substantially enhanced by many-body effects.
[1] Pourovskii LV, Mravlje J, Georges A, Simak SI, Abrikosov IA, arXiv:1603.02287v3
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–19–
INVESTIGATION OF MAGNETOCALORIC EFFECT IN CORRELATED
METALLIC SYSTEMS
Igoshev P.A.1,2, Kokorina E.E.2, Lei Xue3, Nekrasov I.A.2
1Institute of Metal Physics UB RAS, Ekaterinburg, Russia 2Institute of Electrophysics UB RAS, Ekaterinburg, Russia
3Ural Federal University, Ekaterinburg, Russia
In this work we systematically investigated change of entropy S within magnetocaloric
effect for ferromagnetic correlated metallic systems with van Hovwe singularities in the bare
spectra. It was done in the frame of single band Hubbard model within mean-field
approximation. For this model the expression for S can be obtained analytically. Analysis of
the solution of the model shows that in contrast to the Heisenberg model here S strongly
depends not only on spin value and Curie temperature but also on bare electronic structure. This
fact gives additional possibilities to make magnetocaloric effect stronger.
Numerical calculations of S were done for different values of model parameters: small
enough Coulomb interaction U, hopping integrals t and t' and occupancies n. All calculations
were performed for the infinite dimensions Bethe lattice. It is shown that for the case of second
order magnetic phase transition the change of entropy S is always negative for any set of model
parameters. Maximum value of S naturally appearing near Curie temperature is found to
become even stronger while approaching van Hove singularity.
Also in this work magnetocaloric effect is studied for the case of first order magnetic
phase transition. Phase separation region was found as a function of U and n for different t'/t
ratios. Interestingly that in contrast to the second order magnetic phase transition the S value
changes sign in the phase separation region with U or n change. This theoretically allows one
some practical application as, for example, thermo stabilizing system.
To overcome mean-field approximation restrictions magnetocaloric change of entropy
DS was also investigated within dynamical mean-field theory (DMFT).
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–20–
QUANTUM MANY-BODY THEORY AT THE TWO-PARTICLE LEVEL
Alessandro Toschi
Institute of Solid State Physics, TU Wien, Austria
Our physical understanding is mostly based on the quantum many - body
description at the one or at the two-particle level. For strong correlations, the dynamical mean-
field theory (DMFT), a self-consistent approach at the one-particle level, has represented a big
step forward in the last two decades. Recently, however, the scientific frontier has moved to the
treatment of correlations at the two-particle level [1]. This represents a key progress to
understand spectroscopic experiments beyond photoemission [2] and to correctly capture
magnetic screening processes in correlated metals [2,3]. Furthermore, it provides new tools [4]
for a quantitative identification of the fluctuations responsible of characteristic features of
correlated spectral functions, such as, e.g., the pseudogap in the cuprates. Finally, it allows to
study non-local correlations on all length-scales through diagrammatic extensions of DMFT,
such as the dynamical vertex approximation (DΓA) [5]. By means of DΓA, the "fate"
of the Mott-transition in two dimensions [6] and the critical exponents of the Hubbard model in
three dimensions can be calculated [7], opening promising perspectives [8] for non-perturbative
treatments of quantum phase transitions.
G. Rohringer, A. Valli, and A. Toschi, Phys. Rev. B 86, 125114 (2012);
T. Schäfer, et al., Phys. Rev. Lett. 110, 246405 (2013).
A. Toschi, et al., Phys. Rev. B 86, 064411 (2012).
A. Hausoel, et al., submitted; A. Galler, et al., Phys. Rev. B 92 205132 (2015).
O. Gunnarsson et al., Phys. Rev. Lett. 114, 236402 (2015); Phys. Rev. B (2016), in press.
A. Hausoel, et al., submitted; A. Galler, et al., Phys. Rev. B 92 205132 (2015).
A. Toschi, A. A. Katanin, and K. Held, Phys. Rev. B 75 045118 (2007).
T Schäfer, et al., Phys. Rev. B 91, 125109 (2015).
G. Rohringer, A. Toschi, A. Katanin, and K. Held, Phys. Rev. Lett. 107, 256402 (2011).
T.Schäfer, A. Katanin, K. Held, and A. Toschi, arXiv:1605.06355, submitted.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–21–
IMPACT OF NON-LOCAL CORRELATIONS OVER DIFFERENT
ENERGY SCALES: A DYNAMICAL VERTEX APPROXIMATION
STUDY Georg Rohringer 1,2*, Alessandro Toschi 1
1 Institute of Solid State Physics, Vienna University of Technology, Austria
2 Russian Quantum Center, Russia
In this work [1], we investigate how non-local correlations affect selectively, the
physics of correlated electrons over different energy scales, from the Fermi level to the
band-edges. This goal is achieved by applying a diagrammatic extension of dynamical
mean field theory (DMFT), the dynamical vertex approximation (DA) [2,3], to study
several spectral and thermodynamic properties of the unfrustrated Hubbard model in two
and three dimensions. Specifically, we focus first on the low-energy regime by computing
the electronic scattering rate and the quasiparticle mass renormalization for decreasing
temperatures at a fixed interaction strength. This way, we obtain a precise characterization
of the several steps, through which the Fermi-liquid physics is progressively destroyed by
non-local correlations. Our study is then extended to a broader energy range, by analyzing
the temperature behavior of the kinetic and potential energy, as well as of the corresponding
energy distribution functions. Our findings allow to identify a smooth, but definite
evolution of the nature of non-local correlations by increasing interaction: They either
increase or decrease the kinetic energy w.r.t. DMFT depending on the interaction strength
being weak or strong respectively. This reflects the corresponding evolution of the ground
state from a nested-driven (Slater) to a superexchange-driven (Heisenberg) antiferromagnet,
whose fingerprints are, thus, recognizable in the spatial correlations of the paramagnetic
phase. Finally, a critical analysis of our numerical results of the potential energy at the
largest interaction allows us to identify possible procedures to improve the ladder-based
algorithms adopted in the dynamical vertex approximation.
Rohringer G, Toschi A. arXiv: 1604.08748.
Toschi A, Katanin AA, Held K. Phys Rev B 2007; 75:045118.
Rohringer G, Toschi A, Katanin AA, Held K. Phys Rev Lett 2011; 107:256402.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–22–
MODELING OF THE IMPURITY DYNAMICS IN ULTRACOLD ATOMIC
MEDIA V.V. Vyborova 1,2, Y.E. Shchadilova 2,3, A.N. Rubtsov 1,2,4*
1 Moscow State University, Russia 2 Russia Quantum Center, Russia
3 Harvard University, USA 4 VNIIA, Russia
Polaronic problem, describing a massive impurity interacting with a bosonic field, was
first introduced in the context of electron-phonon interaction over 80 years ago. It appears
in many branches of physics, including the black-hole evolution, neutron stars, and many
condensed matter applications. A particular interest arises for polarons formed in artificial
media, such as ultracold atomic gases. These systems allows a sudden change of their
parameters, thus making a problem of the dynamics of polaron formation experimentally
relevant.
We present our studies of the polaron formation dynamics based on the dynamical
extension of the variational Feynman approach. In Feynman approach, the effect of the
bosonic bath is taken into account using the single Gaussian degree of freedom, which is
linearly coupled with the impurity. Parameters of the impurity are chosen according to the
Feynman's variational principle. This approach can be extended to a more general effective
bath, being an arbitrary Gaussian system with the Greens function G (that means an infinite
number of the effective degrees of freedom). Feynman's variational principle yields then
the condition <S>0/G-1=-G, where the average is over the trial Gaussian ensemble.
Moreover, the same equation can used to describe the real-time dynamics, as it follows
from the Schwinger Dyson equations.
For a Frohlich Hamiltonian, we end up with a simple analytical expression for a self-
energy. It allows us to roll out a self-consistent numerical procedure for the polaron
formation dynamics. We will present the time-dependence of the polaron mobility and its
effective mass. Possible auto-localization will be considered as well.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–23–
ELECTRONIC PROPERTIES ASSOCIATED WITH SPIN-ORBIT
COUPLING AND BROKEN SYMMETRY
Tamio Oguchi
The Institute of Scientific and Industrial Research,
Osaka University
Several peculiar electronic properties emerge from spin-orbit coupling (SOC). Rashba effect
is known to be of SOC-origin spin-splitting mechanism by an electric filed. Magnetoelectric
(ME) effect in multiferroics is a coupling between magnetic and electric degrees of freedom
often arising from SOC. The most important clue to the understanding of such SOC-driven
phenomena lurks in the symmetry of the system. In this talk, I will present our recent theoretical
studies on Rashba effect at surfaces and ME effect in non-polar oxides.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–24–
ORIGIN AND MICROSCOPIC MECHANISMS OF
MAGNETOELECTRIC COUPLING IN MULTIFERROIC MANGANITES
Igor Solovyev 1*
1 National Institute for Materials Science, Tsukuba, Japan
I will discuss key mechanisms of the magnetic inversion symmetry breaking and
magnetoelectric coupling in several prototypical types of multiferroic manganiets, originating
from the interplay between competing interatomic magnetic interactions and intraatomic Hund's
coupling. One important aspect of multiferroic research is the correct determination of the
magnetic ground state, which can be highly nontrivial. Particularly, I will argue that the spin-
spiral order, which is frequently anticipated in orthorhombic and some other compounds, is
strongly deformed by relativistic interactions and this deformation is primarily responsible for
the ferroelectric activity in these systems [1]. This analysis is based on the mean-field solution of
the low-energy electron model, derived from the first-principles calculations in the Wannier
basis. Another important question is the coexistence of ferroelectricity and ferromagnetism [2].
In this respect, I will show that an antiferromagnetic order, that breaks the inversion symmetry,
gives rise not only to the ferroelectric activity but also induces finite Dzyaloshinskii-Moriya
interactions, which can lead to the weak ferromagnetism. Such situation is expected in BiMnO3
[3]. Finally, I will present a microscopic model, which captures basis aspects of the
magnetoelectric coupling in manganites. This model is based on the Berry phase theory of the
electronic polarization, where the asymmetric spin-dependent change of the occupied Wannier
functions in evaluated in the framework of the double exchange theory [4]. Particularly, this
model suggests that for an arbitrary noncollinear magnetic structure in orthorhombic
manganites, propagating along the b axis and antiferromagnetically coupled along the c axis, the
polarization can be obtained by scaling the one of the E-type antiferromagnetic phase with the
prefactor depending only on the relative directions of spins and being the measure of the spin
inhomogeneity.
[1] Solovyev IV. Phys Rev B 2011; 83:054404.
[2] Solovyev IV. J Phys Condens Matter 2008; 20:293201.
[3] Solovyev IV, Phys Rev B 2015; 90:024417.
[4] Solovyev IV, Nikolaev SA. Phys Rev B 2014; 90:184425.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–25–
METHODS FOR CALCULATION AND ANALYSIS OF THE
DZYALOSHINSKII-MORIYA INTERACTION
Vladimir Mazurenko 1*
1 Ural Federal University, Russia
The Dzyaloshinskii-Moriya interaction (DMI) plays a crucial role in formation of complex
magnetic states of correlated materials, such as weak ferromagnetism in antiferromagnets, spin
spirals, topologically protected spin textures (skyrmions) in metallic ferromagnets and others.
The determination of this magnetic interaction taking into account hybridization, correlation and
spin-orbit coupling effects is a complex methodological and computational problem requiring a
whole arsenal of numerical techniques.
In my talk, I will discuss different approaches for calculating the Dzyaloshinkii-Moriya
interaction. For instance, the combination of the superexchange theory proposed in the seminal
work by T. Moriya1 and local density approximation taking into account the spin-orbit coupling
gives reliable results in the case of the one-band systems such as low-dimensional cuprates2,3
and nanomaterials with sp-electrons magnetism. For the multi-band systems with different
strength of the spin-orbit coupling and correlation effects the problem of the realistic simulations
of DMI can be solved by using the correlated band method proposed in work4. A special focus
of the talk will be on the analysis of the signs of the orbital contributions to the total anisotropic
exchange interaction.
[1] Moriya T. Phys Rev 1960; 120:91.
[2] Mazurenko VV, Skornyakov SL, Anisimov VI, Mila F. Phys Rev B 2008; 78:195110.
[3] Badrtdinov DI, Volkova OS, Tsirlin AA, Solovyev IV, Vasiliev AN, Mazurenko VV.
arXiv:1604.03333.
[4] Katsnelson MI, Kvashnin YO, Mazurenko VV, Lichtenstein AI. Phys Rev B 2010;
82:100403.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–26–
COHERENT POTENTIAL APPROXIMATION FOR STRONGLY
CORRELATED SYSTEMS WITH SPIN-ORBIT COUPLING
Michael A. Korotin
M.N. Mikheev Institute of Metal Physics, 620990 Ekaterinburg, Russia
The method for calculating the electronic structure of nonstoichiometric and
superstoichiometric compounds with strong electron correlations and spin-orbit coupling is
developed based on the coherent potential approximation. Coherent potential approximation
implemented in the formalism of temperature Green's functions in the basis of localized Wannier
functions. Principles of the method, including the method of accounting for the strong
correlations could be found in [1]. Spin-orbit interaction is as set forth in [2]. The method of
calculation of the parameter ΔV, which describes the characteristics of the potential difference
between the impurity atom and atom replaced by impurity, is discussed also.
As an example, Fig 1 demonstrates the energy dependence of calculated coherent potential
of s-states of an effective oxygen site in TiO2-δ. See [3] for details.
Fig. 1 Self-consistent coherent potential of s-states of effective oxygen site in dependence of
Matsubara frequencies (left) and of real energies (right) for nonstoichiometric rutile TiO2-δ.
Korotin MA, Pchelkina ZV, Skorikov NA, Kurmaev EZ, Anisimov VI. J. Phys.: Condens.
Matter 2014; 26:115501.
Korotin MA, Skorikov NA, Skornyakov SL, Shorikov AO, Anisimov VI. JETP Letters
2014; 100:823.
Korotin MA, Skorikov NA, Zainullina VM, Kurmaev EZ, Lukoyanov AV, Anisimov VI.
JETP Letters 2012; 94:806.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–27–
INTRINSIC TRANSPORT PROPERTIES OF
MONOLAYER BLACK PHOSPHORUS
Alexander N. Rudenko1*
1 Radboud University, Institute for Molecules and Materials,
Heijendaalseweg 135, 6525AJ, Nijmegen, The Netherlands
A few-layer black phosphorus (BP) is a novel 2D semiconductor with strongly anisotropic
electronic properties. Here, we address the problem of intrinsic mobility of single-layer BP
considering charge carrier scattering involving single- and two-phonon processes. We develop a
theory for phonon scattering in anisotropic 2D semiconductors and apply it to study intrinsic
transport in monolayer BP, for which relevant parameters we determine from first-principles
calculations [1]. We show that in contrast to graphene, where two-phonon processes due to the
scattering by flexural phonons dominate at any practically relevant temperature, two-phonon
scattering in BP is considerably less important compared to the single-phonon scattering
involving in-plane modes. This behavior is mainly attributed to a significant difference between
the elastic properties of graphene and BP. As a consequence, phonon-scattering in BP can hardly
be suppressed by depositing BP samples on substrates or by encapsulation. We also find that at n
= 1013 cm-2 and T = 300 K electron mobility in BP is significantly more anisotropic (μxx/μyy ~
6.2) than hole mobility (μxx/μyy ~ 1.4). In the same temperature and doping regime, absolute
values of μxx do not exceed 250 (700) cm2V-1s-1 for holes (electrons), which can be considered as
an upper limit for the mobility in BP at room temperature. Given that these values can be
considerably reduced by other intrinsic and extrinsic scattering mechanisms, the application of
BP as a high mobility semiconductor might be hindered.
[1] A.N. Rudenko, S. Brener, and M.I. Katsnelson, PRL 116, 246401 (2016).
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–28–
AB INITIO EVALUATION AND EXPERIMENTAL VERIFICATION OF
MAGNETIC EXCHANGE PARAMETERS IN INSULATORS
Alexander A. Tsirlin1*
1 Experimental Physics VI, Center for Electronic Correlations and Magnetism, Institute
of Physics, University of Augsburg, Germany
Magnetism of real materials requires multiple parameters for a quantitative microscopic
description. While Heisenberg exchange J is often sufficient for predicting the type of magnetic
ground state, directions of the magnetic moments relative to the crystal axes are determined by
smaller anisotropy terms. In this talk, I will show how both isotropic and anisotropic exchange
couplings can be obtained from DFT-based band-structure calculations and verified by
experimental data, such as magnetization measurements and neutron scattering. Applications to
several frustrated magnets will be presented:
In kagome francisites Cu3Bi(SeO3)2O2X (X = Cl and Br) [1], Dzyaloshinsky-Moriya
interactions remove the extensive ground-state degeneracy induced by the frustrated kagome
geometry, and stabilize ferrimagnetic order, which is uncommon for cuprate compounds. The
magnetization process of francisites is highly anisotropic and conveys information about
anisotropic magnetic interactions in this system.
In Li2NiW2O8 [2], a sequence of commensurate and incommensurate magnetically ordered
phases is observed. This peculiar behavior is rationalized assuming a combination of the
magnetic frustration on the triangular spin lattice and a strong single-ion anisotropy driven by
geometrical distortions of NiO6 octahedra.
[1] Rousochatzakis I, Richter J, Zinke R, Tsirlin AA. Phys Rev B 2015; 91:024416.
[2] Ranjith KM, Nath R, Majumder M, Kasinathan D, Skoulatos M, Keller L, Skourski Y,
Baenitz M, Tsirlin AA. arXiv:1603.01811.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–29–
ELECTRONIC CORRELATIONS AND TOPOLOGICAL FERMI
SURFACE TRANSITION IN THE IRON-BASED CHALCOGENIDES
Sergey Skornyakov1*, Ivan Leonov 2, Vladimir Anisimov 1, Dieter Vollhardt 2
1 Institute of Metal Physics Ural Branch RAS, Russia
2 Theoretical Physics III, Center for Electronic Correlations and Magnetism, University
of Augsburg, Germany
We present results of a theoretical investigation of the electronic structure and phase stability
of the parent chalcogenide compound FeSe obtained within a combination of density functional
theory and dynamical mean-field theory [1]. Our results reveal an entire reconstruction of the
Fermi surface upon a moderate expansion of the lattice (Lifshitz transition), with a change of
magnetic correlations from the in-plane magnetic wave vector (π,π) to (π,0). We attribute this
behavior to a correlation-induced shift of the van Hove singularity originating from the xy and
xz/yz bands at the M-point across the Fermi level. We predict an isostructural transition of FeSe
upon a hydrostatic and uniaxial expansion of the lattice volume as well as a topological change
of the Fermi surface of FeSe upon partial substitution Se by Te, which is accompanied with a
sharp increase of the local moments. We expect that these changes are responsible for the
experimentally observed increase of the critical temperature in FeSe upon doping with Te. The
microscopic origin for superconductivity in this system is then due to a van Hove singularity
close to the Fermi level. This identification may open a new route to increase Tc even further.
[1] Leonov I, Skornyakov SL, Anisimov VI, Vollhardt D. Phys Rev Lett 2015; 115:106402.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–30–
FUNCTIONAL ANTIFERROMAGNETIC MATERIALS FOR
SPINTRONICS APPLICATIONS: CHALLENGE FOR AB INITIO
COMPUTATIONS
Sergii Khmelevskyi
Center for Computational Materials Science,
Vienna University of Technology, Austria
The antiferromagnetic materials become an ultimate importance in the modern electronics
since the discovery of the GMR effects. They provide a pining of the ferromagnetic layers and
being an integral part of almost any computer memory devices. Since the recent development
related to the possibility of the laser ultrafast switching of the magnetization in ferri- and
antiferromagnets (AFM), finding new routes for application of the spin-orbit coupling effects in
spintronic and abundance of the critical elements on the market – currently the search and design
of new AFM materials for applications, with stringent technological requirements on their
properties, is one of the main stream of the development in magnetic material science. In this
talk I will give an overview of the subject and illustrate a major role that the first-principles
modeling have in the AFM material development. The discovery of the new high-temperature
AFM materials, the funding of new routes in spintronics using ab initio modeling, application of
the magnetic force theorem for predicting the Neél temperature and local anisotropies in
functional AFM alloys on real examples (Mn2Au, Ru2MnX, V3Al, Mn3Ga, binaries
Mn(Ir,Pd,Ni) etc.) will be presented and discussed.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–31–
THE ROLE OF INTERFACES FOR STRUCTURAL
TRANSFORMATIONS AMONG AUSTENITE, FERRITE AND
CEMENTITE IN Fe-C ALLOYS
Tilmann Hickel, Xie Zhang, Jutta Rogal, Jörg Neugebauer
Max-Planck-Institut für Eisenforschung, Düsseldorf, Germany
During the cooling process of Fe-C alloys a decomposition of the high-temperature
austenitic phase into ferrite and cementite takes place. The atomistic modelling of the involved
processes at the interface is challenged by the simultaneous changes in Fe lattice and
redistribution of C to accommodate the reduced C solubility.
With a combination of the orientation relationships between austenite, ferrite and cementite,
we identify an intermediate structure, which serves as a link between the three phases. It is
extended over a few atomic layers and stabilizes the interfaces similar to complexions. Based on
this framework, different mechanisms depending on the local conditions (C concentration,
strain, magnetism) are revealed from ab initio nudged elastic band simulations, which allow us
to construct a theory for the structural transformations among austenite, ferrite, cementite and
martensite.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–32–
APPLICATION OF THE AMULET CODE FOR DFT+DMFT CALCULATIONS
OF REALISTIC COMPOUNDS
Alexander Poteryaev1
1Institute of Metal Physics, 620990, Ekaterinburg, Russia
Over last twenty years, the DFT+DMFT method has successfully applied for a description
of different properties of strongly correlated materials [1,2]. This combination of the density
functional theory (DFT) to characterize material specific aspect of problem and the dynamical
mean field theory (DMFT) to treat strong electronic correlations allows one to understand a
physics of many systems. In this hands-on, we present Advanced Materials simULation
Ekaterinburg's Toolbox (AMULET [3]) - a suite of computer codes for ab initio DFT+DMFT
calculations. Electronic, magnetic and structural properties of realistic strongly correlated
compounds or alloys can be easily investigated with this instrument. During the session, the
DFT+DMFT and CPA+DMFT approaches will be considered in brief, while the main focus will
be on practical aspects. Participants will learn how AMULET interfaces with different band
structure packages. In a practical computer based part of hands-on, they will get skills on setting
up input files, running code and understanding results. Particularly, we consider in details the
FexNi1-x alloy [4] and calculate magnetic properties of this compound as function of
concentration.
[1] Anisimov V., Poteryaev A., Korotin M., Anokhin A., and Kotliar G., Journal of Physics:
Condensed Matter 9, 7359 (1997).
[2] Lichtenstein A. and Katsnelson M., Physical Review B 57, 6884 (1997).
[3] www.amulet-code.org
[4] Poteryaev A., Skorikov N., Anisimov V., and Korotin M., Physical Review B 93, 205135
(2016).
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–33–
AB INITIO THERMODYNAMIC DESCRIPTION OF ADVANCED
STRUCTURAL MATERIALS: STATUS AND CHALLENGES
Jörg Neugebauer, Albert Glensk, Fritz Koermann, Blazej Grabowski
and Tilmann Hickel
Max-Planck-Institut für Eisenforschung, Düsseldorf, Germany
Modern engineering materials have evolved from simple single phase materials to nano-
composites that employ dynamic mechanisms down to the atomistic scale. The structural and
thermodynamic complexity of this new generation of structural materials presents a challenge to
their design since experimental trial-and-error approaches as successfully used in the past are
often no longer feasible. Ab initio approaches provide perfect tools to new design routes but face
serious challenges: Free energies of the various phases are almost degenerate, requiring
theoretical formalisms that accurately capture all relevant entropic contributions due to
electronic, vibrational or magnetic excitations, as well as their coupling such as phonon-phonon,
magnon-phonon interactions or spin-quantization. In addition, their hierarchical nature with
respect to length and time makes them challenging for any atomistic approach. Combining
accurate first principles calculations with mesoscopic/macroscopic thermodynamic and/or
kinetic concepts allows us now to address these issues and to determine free energies and
derived thermodynamic quantities that often rival available experimental data. The flexibility
and the predictive power of these approaches but also their present limitations will be discussed
for examples ranging from modern ultra-high strength steels to light weight metallic alloys.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–34–
FINITE TEMPERATURE EFFECTS IN AB INITIO SIMULATIONS OF
ALLOY THERMODYNAMICS
Igor Abrikosov
Department of Physics, Chemistry and Biology (IFM), Linköping University,
SE-581 83 Linköping, Sweden
Ab initio electronic structure theory is known as a useful tool for prediction of materials
properties, for their understanding, as well as for determination of parameters employed in
higher-level modeling. However, majority of simulations still deal with calculations in the
framework of density functional theory (DFT) with local or semi-local functionals carried out at
zero temperature. In this talk, we present new methodological solutions, which go beyond this
approach and explicitly take into account finite temperature effects. Basic ideas behind novel
techniques for first-principles theoretical simulations of lattice dynamics, as well as their
coupling to systems with magnetic excitations and configurational disorder are introduced. The
capabilities of the Temperature Dependent Effective Potential (TDEP) method [1], the
Disordered Local Moment Molecular Dynamics (DLM-MD) [2], and the combined technique
[3] are demonstrated in applications for Ti-based alloys, iron carbides, transition metal nitrides
and their alloys [4].
O. Hellman, I. A. Abrikosov, and S. I. Simak, Phys. Rev. B 84, 180301(R) (2011); O.
Hellman, P. Steneteg, I. A. Abrikosov, and S. I. Simak, Phys. Rev. B 87, 104111 (2013);
O. Hellman and I. A. Abrikosov, Phys. Rev. B 88, 144301 (2013).
P. Steneteg, B. Alling, and I. A. Abrikosov, Phys. Rev. B 85, 144404 (2012).
N. Shulumba, B. Alling, O.Hellman, E. Mozafari, P. Steneteg, M. Odén, and
I. A. Abrikosov, Phys. Rev. B 89, 174108 (2014).
N. Shulumba, O. Hellman, L. Rogström, Z. Raza, F. Tasnadi, I. A. Abrikosov, and M.
Odén, Appl. Phys. Lett. 107, 231901 (2015).
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–35–
WEYL SEMI-METAL: A NEW TOPOLOGICAL STATE
IN CONDENSED MATTER
Sergey Savrasov
University of California Davis, USA
Using first-principles electronic structure calculations we investigate novel phases that
emerge from the interplay of electron correlations and strong spin-orbit coupling [1]. We focus
on describing the topological semimetal, a three-dimensional phase of a magnetic solid, which is
a three-dimensional analog of graphene with linearly dispersing excitations. This state provides
a condensed-matter realization of Weyl fermions that obeys a two-component Dirac equation. It
also exhibits remarkable topological properties manifested by surface states in the form of Fermi
arcs, which are impossible to realize in purely two-dimensional band structures. We discuss that
it may be realized in a class of pyrochlore iridates (such as Y2Ir2O7) based on calculations using
the LDA +U method and overview some recent experimental discoveries of Weyl semimetal
materials.
Xiangang Wan, Ari Turner, Ashvin Vishwanath, Sergey Y. Savrasov, Phys. Rev. B 83,
205101 (2011).
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–36–
LOCALLY DESTROYED CRYSTAL ORDER IN Ti-Fe ALLOYS
D. W. Boukhvalov1, Yu. N. Gornostyrev,2,3 and M. I. Katsnelson4
1Department of Chemistry, Hanyang University, Seoul, Korea. 2Institute of Metal Physics, UB of RAS, Ekaterinburg, Russia 3Institute of Quantum Materials Science, Ekaterinburg, Russia
4Radboud University, Institute for Molecules and Materials, Nijmegen, Netherlands
Titanium-based alloys of transition metals demonstrate unusual (for metallic systems)
properties such as negative temperature coefficient of resistivity, pseudogap in infrared optical
spectra, strong concentration anomalies of sound velocities and attenuation, etc. We present the
results of ab initio modeling of structure of Ti-Fe, a typical representative of quenched Ti-based
transition-metal alloys. We have demonstrated that beyond the solubility limit this alloy cannot
be described in common terms of substitutional and interstitial alloys. Instead, very stable local
clusters are formed in both hcp and bcc matrices, with almost identical structures. This gives an
example of geometrically frustrated state and explains unusual concentration behavior of
Mössbauer spectra discovered long ago for this system.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–37–
TEMPERATURE-DRIVEN MARTENSITIC PHASE TRANSITIONS
FROM FIRST PRINCIPLES
Sergei I. Simak 1*
1 IFM, Linköping University, SE-58183 Linköping, Sweden
We discuss how a martensitic phase transition can be studied from first principles in the
framework of molecular dynamics, which allows for a proper account of temperature. As an
example the martensitic phase transition in the shape-memory alloy NbRu is considered. Its
thermophysical properties are reproduced in good agreement with experiment and the effect of
chemical disorder on the mechanical stability is elucidated.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–38–
IMPURITY SEGREGATION AND ITS EFFECT ON THE GRAIN
BOUNDARY EMBRITTLEMENT IN TI: EFFECTS OF CHEMICAL AND
STRUCTURAL CONTRIBUTIONS.
Vsevolod Razumovskiy, D. Scheiber, L. Romaner
Materials Center Leoben Forschung GmbH (MCL), Austria
In this paper we perform a series of density functional theory (DFT) calculations to
investigate segregation of 3d, 4d and 5d alloying elements to grain boundaries (GB) in bcc, fcc
and hcp Ti. We study the effect of these elements on GB cohesion and analyze the role of
structural and chemical contributions to the GB strengthening energy. The free surface and GB
segregation energies are analyzed by comparing results of DFT calculations to results of existing
physical segregation models. Following the concept of low-alloying additions proposed in Ref
[1], we suggest a list of the most promising alloying elements from the point of view of GB
cohesion enhancement in bcc, fcc and hcp Ti.
[1] V.I. Razumovskiy, A. Y. Lozovoi, I. M. Razumovskii Acta Mater. 82 (2015) 369-377
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–39–
THE INTERACTION OF HYDROGEN INTERSTITIALS WITH
GRAIN BOUNDARIES IN BCC IRON Anastasiya Verkhovykh, Alexander Mirzoev*
South Ural State University, Russia
Hydrogen that is accumulated within the grain boundaries (GB) can lead to a decrease of the
critical strain required to fracture the material. The paper presents result of modeling of
hydrogen–grain boundary interaction in ferromagnetic bcc iron. Modeling was performed using
density functional theory with generalized gradient approximation (GGA’96), as implemented in
WIEN-2k package. Three fully relaxed tilt grain boundaries, Σ5(310), Σ5(210) and Σ3(111),
were studied. The supercells contained 40–48 atoms, i.e. 20–24 atoms in each of the two
‘grains’. The cohesive energy CE and binding energy
BE of hydrogen to that determines the
strengthening or embrittling effect can be defined as follows: H H
C GB GB fs fsE E E E E (1) H H
B GB GB bulk bulkE E E E E (2)
where EGB and H
GBE are the total energies of the impurity-free GB supercell and of the same
supercell with one H interstitial, Efs and H
fsE are the total energies of the free surface supercell
and with one H interstitial, Ebulk and H
bulkE are the energies of the commensurate Fe bulk supercell
and with one H interstitial. The results of calculation by the formulas (1)- (2) shown in Table 1.
Table 1. The cohesive (ΔEC) and the binding energy (ΔEB) of hydrogen to grain boundary.
GB types ΔEC (eV) ΔEB (eV)
Our results Other
results
Our results Other
results
Σ5(310) 0,68 - 0,43 0,4 [3]
Σ5(210) 0,07 - 0,81 -
Σ3(111) 0,41 0,31[2] 0,39 0,49 [1]
Exp. - 0,51 [4]
This work was supported by Russian Science Foundation (grant №16-19-10252) and RFFI (№
16-03-00486).
Matsumoto R. et al. Prog. Nucl. Sci. Technol. 2011; 2: 9.
Tian Z.X. et al. J. Phys., Condens. Matter 2011; 23: 015501.
Du Y.A. et al. Phys. Rev. B 2011;84: 144121.
Ono K. and Meshii M. Acta Metal Mater. 1992;40:1357.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–40–
AB INITIO SIMULATION OF PHOSPHORUS IN BULKS, AT SURFACES
AND INTERFACE OF FCC Fe AND κ-CARBIDE
Nadezhda Medvedeva
Institute of Solid State Chemistry, Ural Branch RAS, Ekaterinburg, Russia
First-principles atomistic methods provide reliable information on the microscopic
mechanisms governing the phase stability, impurity partitioning as well as deformation behavior
of iron alloys with carbide precipitates. Much attention is paid to the mechanisms of fracture and
plasticity, which may be studied using ab initio calculations of cleavage and shear
characteristics. The modeling of alloying effect on the interfacial structure and bonding of
carbide/Fe is very important to elucidate the dislocation pinning mechanism. The knowledge of
interfacial bonding is important to predict characteristics of precipitation (nucleation, growth and
coarsening) as well as the embrittling potency of impurities. Ab initio density functional calculations were performed to study phosphorus effect on the
structural, electronic and magnetic properties of bulk, surface and interface of κ-carbide
(Fe3AlC) and fcc Fe. The aim of this study was to shed light on its behavior in austenitic alloys
with the κ-carbide particles, where phosphorus promotes both intergranular and transgranular
embrittlement. The binding energies were calculated for phosphorus in the substitutional and
interstitial positions in bulks/surfaces/interface to predict its stable occupation sites. Our results
indicate that P atoms occupy the substitutional positions in both bulks and repel each other in
fcc Fe, whereas they show a trend to ordering in κ-carbide that favors its formation. Phosphorus
in κ-carbide reduces sharply its cleavage characteristics. The microscopic mechanism of
phosphorus effect on intergranular cleavage was related to strong anisotropy of Fe-P bonds
leading to the appearance of large structural voids in κ-carbide. The interstitial octahedral sites
are most stable for phosphorus adsorption at the (001)Fe3AlC and (001)fcc Fe surfaces, whereas
substitution for Fe is preferable at the (001)Fe3AlC/(001)fcc Fe interface. A strong tendency for
phosphorus segregation at surface explains the embrittling transgranular behavior of
phosphorus.
N.I.M acknowledges the support from the Russian Foundation for Basic Research (Grant 14-03-
00324а)
Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–41–
EFFECT OF COMPOSITION ON ANTI-PHASE BOUNDARY
ENERGY IN NI3AL BASED ALLOYS
Oleg I. Gorbatov1*, I. L. Lomaev1,2, Yu. N. Gornostyrev1,2
1 Institute of Quantum Materials Science, Russia
2 Institute of Metal Physics Ural Branch RAS, Russia
The effect of composition on the anti-phase boundary (APB) energy of Ni based L12-ordered
alloys is investigated by ab initio calculations employing the coherent potential approximation
[1]. The calculated APB energies for {111} and {001} planes reproduce experimental values of
the APB energy. The APB energies for the non-stoichiometric γ'-phase increase with Al
concentration and are in line with the experiment. The magnitude of the alloying effect on the
APB energy correlates with the variation of the ordering energy of the alloy according to the
alloying element's position in the 3d row. The elements from the left side of the 3d row increase
the APB energy of the Ni based L12-ordered alloys, while the elements from the right side
slightly affect it except Ni.
[1] Gorbatov OI, Lomaev IL, Gornostyrev YuN, Ruban AV, Furrer D, Venkatesh V, Novikov
DL, Burlatsky SF. Phys. Rev. B 93, 224106 (2016).
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–42–
AB INITIO MODELING FOR UNDERSTANDING AND PREDICTING
NOVEL ALLOY BEHAVIOR
James R. Morris1,2
1Materials Science and Technology Division, Oak Ridge National Lab, Oak Ridge, TN 37830 2Materials Science and Engineering Department, University of Tennessee, Knoxville, TN 37996
Ab initio calculations, combined with experiment and also with longer length scale
modeling, provides critical information on the stabilization of particular phases and
microstructures, under various thermal and mechanical stresses. This talk will focus on
approaches for connecting these issues, with a particular focus on high entropy alloys (HEAs)
formed of a large number of near-equiatomic elements. These alloys pose new questions
concerning the ability to predict single phase materials in alloys with a large number of
components, particularly metastable phases. We demonstrate that high-throughput calculations
provide important predictive information as to which compositions may form single-phase solid
solutions, including those where Hume-Rothery considerations have been shown to be
inadequate. From these same calculations, effective Monte Carlo models have been used to
examine phase evolution of Al-containing HEAs that show a complex set of phase
transformations. These same materials exhibit unusual mechanical properties, including the
unusual combination of increased strength and ductility under cooling conditions. The materials
challenge traditional considerations of solid-solution hardening, and atomistic simulations may
provide critical insight into these processes.
This work has been supported by the U. S. Department of Energy, Office of Science, Basic
Energy Sciences, Materials Science and Engineering Division.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–43–
AB INITIO BASED MODELS OF DISORDERED MATERIALS
Pavel A. Korzhavyi 1,2*
1 KTH Royal Institute of Technology, Stockholm, Sweden
2 Institute of Metal Physics Ural Branch RAS, Russia
Ab initio simulations of disordered solids at finite temperatures are becoming an important
part of integrated computational materials engineering. The fundamental ab initio approach
allows one, in principle, to predict the properties of alloys as functions of composition and
temperature, thereby enabling computer-aided design of high-performance materials [1]. In
practice, fully detailed simulations require large computational resources in order to follow the
evolution of a complex system in the available phase space (comprising electronic, magnetic,
vibrational, and compositional degrees of freedom) [2,3]. Therefore it is important to develop
methodologies of coarse-graining, i.e. procedures to extract the most essential information about
the system’s behavior and represent it effectively, using a few leading variables, while
integrating out the rest of the variables as just contributing to the statistics. To keep the
predictive power on the ab initio level, these procedures should be mathematically well-defined
and physically sound [4]. The existing approaches to coarse-graining in the field of finite-
temperature modeling of metallic alloys will be briefly reviewed. Application examples of
effective-medium-based methods to describe the thermal properties of steel and high-
temperature alloys will be given; recent achievements and remaining challenges will be
discussed.
[1] Hickel T, Kattner UR, Fries SG. Phys. Status Solidi B 2014; 251:9.
[2] Körmann F, et al. Phys. Status Solidi B 2014; 251:53.
[3] Abrikosov IA, et al. Current Opinion in Solid State and Materials Science 2016; 20:85.
[4] Schmitz GJ, et al. JOM 2015; 68:70.
*Email: [email protected]
AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–44–
SCALING BEHAVIOR OF THE COMPTON PROFILE OF ALKALI
METAL ELEMENTS
Michael Sekania1,2,*, Wilhelm H. Appelt1,3, Dieter Vollhardt1, Liviu Chioncel 1,3
1 Center for Electronic Correlations and Magnetism,
Institute of Physics, University of Augsburg, D-86135 Augsburg, Germany 2 Andronikashvili Institute of Physics, Tamarashvili 6, 0177 Tbilisi, Georgia
3 Augsburg Center for Innovative Technologies,
University of Augsburg, D-86135 Augsburg, Germany
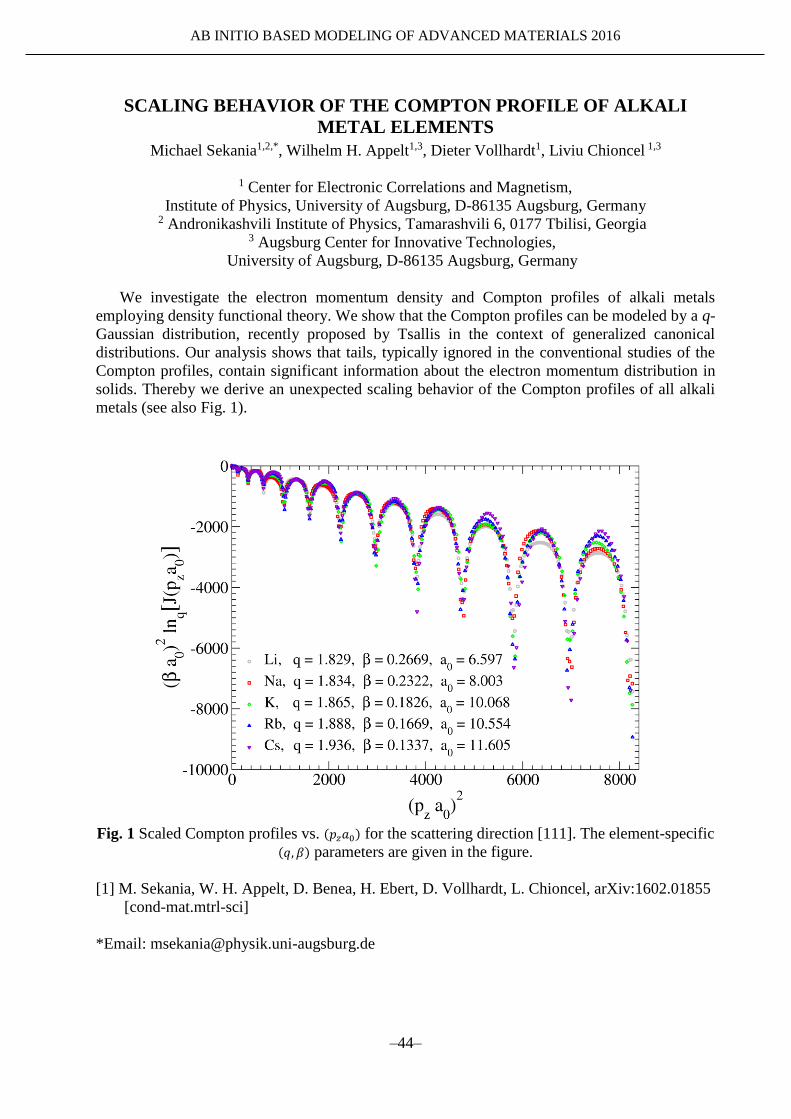
We investigate the electron momentum density and Compton profiles of alkali metals
employing density functional theory. We show that the Compton profiles can be modeled by a q-
Gaussian distribution, recently proposed by Tsallis in the context of generalized canonical
distributions. Our analysis shows that tails, typically ignored in the conventional studies of the
Compton profiles, contain significant information about the electron momentum distribution in
solids. Thereby we derive an unexpected scaling behavior of the Compton profiles of all alkali
metals (see also Fig. 1).
Fig. 1 Scaled Compton profiles vs. (𝑝𝑧𝑎0) for the scattering direction [111]. The element-specific
(𝑞, 𝛽) parameters are given in the figure.
[1] M. Sekania, W. H. Appelt, D. Benea, H. Ebert, D. Vollhardt, L. Chioncel, arXiv:1602.01855
[cond-mat.mtrl-sci]
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–45–
AB INITIO INVESTIGATION OF GRAIN BOUNDARY SEGREGATION
IN ULTRAFINE-GRAINED AL-X [X=Mg,Zn,Si,Cu] ALLOYS Mikhail Petrik 1,2*, Yuri Gornostyrev 1,2
1 Institute of Metal Physics Ural Branch RAS, Russia
2Institute of quantum materials science, Russia
Producing ultrafine-grained (UFG) alloys by severe plastic deformation leads to segregation
of alloying elements at grain boundaries (GB), which considerably affects the material’s
properties. Ab initio calculations provide unique possibility to investigate processes of solute-
GB interactions accounting electronic and deformation mechanisms [1]. To understand main
experimental features of agglomeration formation we obtained ab initio results of interactions of
impurity atoms with the special type GB. Also the effect of GB presence on the solute-solute
interactions has been considered.
It is shown that electronic mechanisms of interaction between solutes prevail in Al-Zn and
Al-Cu alloys while the behavior of Mg solutes can be explained in deformation terms.
Segregation energy calculation showed that Mg atoms prefer to occupy the center of the GB as
well as the vicinity of the GB. Long-distance attractive Mg-Mg interactions near GB region lead
to heterogeneous agglomeration formation. In contrast, Zn atoms prefer to occupy interstitial
positions and are arranged into thin layers along GB. Accompanied with vacancy generation this
effect facilitates grain boundary sliding. Difference in chemical bonding of solute atoms X
[X=Mg,Zn,Si,Cu] with Al and its effect on the ductility of the alloys is discussed [2]. The results
obtained are consistent with experimental observation of super-strength in UFG Al-Mg alloys
and super-ductility in Al-Zn alloys.
L.E. Karkina, I.N. Karkin, A.R. Kuznetsova, , I.K. Razumov, P.A. Korzhavyi, Yu.N.
Gornostyrev Solute–grain boundary interaction and segregation formation in Al: First
principles calculations and molecular dynamics //Computational Materials Science, 112, 18-
26 (2016)
M. V. Petrik, A. R. Kuznetsov, N. Enikeev, Yu. N. Gornostyrev, R. Z. Valiev Ab initio
based analysis of grain boundary segregations in ultra-fine grained Al alloys // to be
published
*Email:[email protected]

ABSTRACTS
POSTERS

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–47–
AB INITIO MODELLING OF DIAMOND-LIKE MATERIALS
Vladimir Greshnyakov*, Evgeny Belenkov
Chelyabinsk State University, Russia
Carbon diamond-like phases are phases consisting of four-coordinated carbon atoms.
Diamond-like phase structures can be theoretically obtained by linking or superpositioning of
precursors which are composed of three-coordinated atoms [1]. These precursors are graphene
layers, carbon nanotubes, fullerene-like clusters and three-dimensional graphites. Nanoclusters
of diamond-like phases have been obtained by this method, the structures of which were
geometrically optimized by semi empirical quantum mechanical methods [1, 2]. In the central
part of these clusters, the least distorted unit cells have been carved. Further, the unit cells were
geometrically optimized by density functional theory methods based on periodic boundary
conditions [3–6].
As a result of first-principles calculations, the possibility of stable existence of thirty-six
diamond-like phases is established (including the cubic diamond) [3–6]. The diamond-like phase
densities are in the range from 35 to 102 % relative to the diamond density. Sublimation
energies of these phases decrease with increasing the degree of structural tension of these phases
relative to the cubic diamond structure. Almost all of the diamond-like phases are wide bandgap
semiconductors with band gaps from 0.9 to 5.6 eV. Carbon diamond-like phases have high
mechanical characteristics: the bulk modulus varies from 141 to 452 GPa; the Vickers hardness
ranges from 49.4 to 90.1 GPa. The analysis of possible synthetic routes of the new phases has
shown that the phase transition of graphite into the cubic diamond or diamond-like phases
occurs at a pressure greater than 58 GPa. The calculated transition pressure value agrees well
with the experimental data (at P > 50 GPa and T < 300 K).
The research was funded by RFBR according to the research project No. 16-33-00030 mol_a.
[1] Greshnyakov VA, Belenkov EA. J Exp Theor Phys 2011; 113:86.
[2] Belenkov EA, Greshnyakov VA. J Struct Chem 2014; 55:409.
[3] Belenkov EA, Greshnyakov VA. Phys Solid State 2015; 57:205.
[4] Belenkov EA, Greshnyakov VA. J Mater Sci 2015; 50:7627.
[5] Belenkov EA, Greshnyakov VA. Phys Solid State 2015; 57:1253.
[6] Belenkov EA, Greshnyakov VA. Phys Solid State 2015; 57:2331.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–48–
DFT AND MULTI-REFERENCE PERTURBATION THEORY
CALCULATIONS OF THE STRUCTURES AND UV-VIS SPECTRA OF
ADAMANTANE-CONTAINING MOLECULES, POTENTIAL
ANTIBACTERIAL AGENTS
Maksim Shundalov 1*, Anna Matsukovich 2, Sergey Gaponenko 1,2
1 Belarusian State University, Minsk, Belarus
2 B.I. Stepanov Institute of Physics, NAS of Belarus, Minsk, Belarus
The incorporation of an adamantyl moiety into several molecules results in compounds with
relatively high lipophilicity, which in turn can modify the biological availability of these
molecules. Several adamantane derivatives were associated antimicrobial and anti-inflammatory
activities.
The structures of N'-(adamantan-2-ylidene)benzohydrazide (C17H20N2O, I, 4 conformers)
[1], 3-(adamantan-1-yl)-4-phenyl-1-[(4-phenylpiperazin-1-yl)methyl]-1H-1,2,4-triazole-5(4H)-
thione (C29H35N5S, II, 3 conformers) [2] and ethyl 4-{[3-(adamantan-1-yl)-4-phenyl-5-
sulfanylidene-4,5-dihydro-1H-1,2,4-triazol-1-yl]methyl}piperazine-1-carboxylate
(C26H35N5О2S, III, 6 conformers) [3] have been obtained at the B3LYP/cc-pVDZ level of
theory. The calculations of the UV-vis spectra for all conformers were performed at the
CASSCF/XMCQDPT2 [4] level of theory. The UV-vis absorption spectra of all compounds for
the ethanol solution were measured in the 200–450 nm region. The UV-vis spectrum (Fig. 1) of
the compound I in the 220–320 nm region reveals two bands that can be explained based on the
ab initio calculations of the “side” trans-conformers of the compound I at the MRPT level of
theory. For the compounds II and III we also obtained a good agreement between experimental
and calculated spectra.
Fig. 1 The absorption spectra of the compound I: experimental in ethanol (1)
and calculated at the CASSCF/XMCQDPT2 level of theory (2)
Almutairi M.S. et al. Acta Cryst. 2012; E68: o2247.
Al-Abdullah E.S. et al. Acta Cryst. 2012; E68: o345.
Al-Abdullah E.S. et al. Acta Cryst. 2012; E68: o531.
Granovsky AA. J. Chem. Phys. 2011; 134: 214113.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–49–
MODELLING OF THE MAGNETIC INTERACTION OF STRONGLY
CORRELATED SYSTEMS
Gerasimov A.O.1*, Mazurenko V.V.1*, Skornyakov S.L.2
1 Ural Federal University, Russia
2 Institute of Metal Physics Ural Branch RAS, Russia
This work is devoted to the implementation of the scheme for the calculation of the magnetic
interactions in strongly correlated materials. It shows unique electronic and magnetic properties
which can be used to create new technology. In such compounds at specific electronic states the
average value of the Coulomb interaction of the same order of magnitude as the kinetic energy,
so the construction of the perturbation theory in one of the parameters is not possible. A method
to solve this problem is - DMFT.
The developed numerical scheme includes:
Carrying out ab initio calculations using LDA;
The construction of the Hamiltonian in Hubbard model and the solution of the
Hamiltonian using numerical method DMFT;
Calculation of magnetic interactions on the basis of results of the DMFT-calculations
(Local forces theorem, see at [1]).
The scheme was tested on the BCC Fe crystal. One of the parameters for getting the
magnetic properties of strongly correlated systems is - the integral of the exchange interaction,
that was calculated (see at Fig.1) using the theorem of local forces [1]:
𝐽𝑖𝑗 = −𝑇𝑟𝜔𝐿(𝛴𝑖𝑆𝐺𝑖𝑗
↑ 𝛴𝑗𝑆𝐺𝑖𝑗
↓ ), (1)
where 𝛴𝑖𝑆 =
1
2(𝛴𝑖
↑ − 𝛴𝑖↓). The calculated Curie temperature with the exchange parameters from
Fig.1 is 966K, that close to that were obtained in [2] and to experimental 1045K [1].
In the future we plan to work to study the properties FeMn alloy.
Fig. 1 Exchange parameters in BCC Fe (in meV) as a function of shells ( = 10,
U = 2.3 eV, J = 0.9 eV).
[1] Katsnelson M. I., Lichtenstein A. I. Phys. Rev. B. 2000; 61:8906.
[2] Kvashnin Y. O., Granas O., Di Marco I. Phys. Rev. B. 2015; 91: 125133.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–50–
EFFECT OF DYNAMICAL ELECTRON CORRELATIONS ON
COLLECTIVE MAGNETIC EXCITATIONS IN CRO2
Ilya Kashin1*, Igor Solovyev 1, 2, Vladimir Mazurenko1
1 Department of Theoretical Physics and Applied Mathematics, Ural Federal University, Russia
2 National Institute for Materials Science, Japan
CrO2 is known to be a rare example of stoichiometric oxide, demonstrating half-metallic
(HM) and ferromagnetic (FM) properties. It is widely used in magnetic recording tape
production and serves as promising candidate for implementation in various spin-dependent
transport phenomena.
In this study we investigate the effect of dynamical electron correlations on the stability of
ferromagnetism in CrO2 by means of considering the collective magnetic excitations, such as
spin waves. For this purpose, we construct the realistic low-energy model for t2g band of Cr
atoms, derived from the first-principles electronic structure calculations, and solve it by means
of unrestricted Hartree-Fock approach (HF) and the dynamical mean-field theory (DMFT).
Obtained on-site Green’s function was used to calculate the interatomic exchange interactions in
the framework of the theory of infinitesimal spin rotations [1], which were combined to find the
value of spin stiffness constant. The results are shown at Fig. 1. Switching the sign of D to
negative in case of DMFT indicates that the strong antiferromagnetic longer-range exchange
interactions destabilize FM ground state. It leads to conclusion that accounting for dynamical
electron correlations is of the great significance to correctly describe the magnetic properties of
CrO2, the canonical and technologically important half-metallic ferromagnet.
Fig. 1 Spin stiffness constant in CrO2
This work is supported by a grant from Russian Foundation for Basic Research (Project № 16-
32-00076).
[1] Katsnelson MI, Lichtenstein AI. Phys Rev B 2000; 61:8906.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–51–
PRESSURE DEPENDENSE OF THE ELECTRONIC STRUCTURS OF
Sr3Ir2O7
Sergei Andreev 1*, Igor Solovyev 1, 2, Vladimir Mazurenko 1
1 Ural Federal University, Russia
2 National Institute for Materials Science, Japan
The iridium oxides with the Ruddlesden-Popper structure (Srn+1 Irn O3n+1) are in the focus of
modern condensed matter physics. It was shown that a strong spin-orbit coupling in these oxides
may lead to insulating state with a very narrow gap in the electronic spectrum [1]. This state can
be destroyed by applying the pressure. For Sr3Ir2O7 a structural transition was detected at a
pressure of 54 GPa at room temperature that is associated with the transition from the tetragonal
to monoclinic symmetry [2]. However, the changes in the spectrum of electron excitations
remain unknown.
The aim of the study is a theoretical description of the electronic and magnetic properties
under pressure. The LDA+U+SO calculations were conducted by using package Quantum-
Espresso. These calculations for the structures at different pressures revealed that the
compression of the unit cell is accompanied by an increase of the in-plane rotation angle of the
IrO6 octahedron, while the Ir-O distance decreases. The insulating state of Sr3Ir2O7 is therefore
very sensitive to the value of the pressure. We find a closure of the electronic gap and a
transition to a metallic state above 20 GPa. Another interesting result is a suppression of the
magnetic moment at high pressure.
This work was supported by grant RSF 14-12-00306.
[1] Witczak-Krempa W, Chen G, Kim YB, Balents L. Annu. Rev. Condens. Matter Phys 2014;
5:57.
[2] Donnerer C, Feng Z, Vale JG, Andreev SN, Solovyev IV, Hunter EC, Hanfland M, Perry
RS, Ronnow HM, McMahon MI, Mazurenko VV, McMorrow. arXiv:1508.04320v1.
*Email: [email protected]
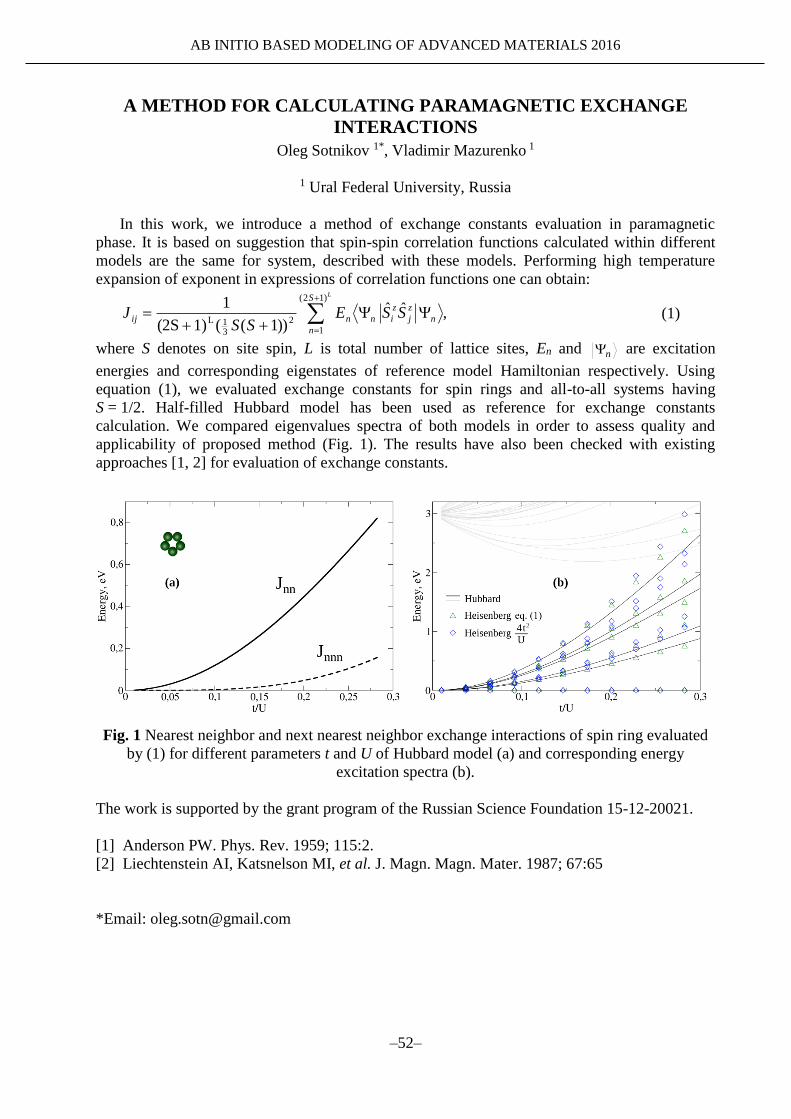
AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–52–
A METHOD FOR CALCULATING PARAMAGNETIC EXCHANGE
INTERACTIONS
Oleg Sotnikov 1*, Vladimir Mazurenko 1
1 Ural Federal University, Russia
In this work, we introduce a method of exchange constants evaluation in paramagnetic
phase. It is based on suggestion that spin-spin correlation functions calculated within different
models are the same for system, described with these models. Performing high temperature
expansion of exponent in expressions of correlation functions one can obtain:
,ˆˆ))1((1)(2S
1 )12(
12
31L
LS
n
n
z
j
z
innij SSESS
J (1)
where S denotes on site spin, L is total number of lattice sites, En and n are excitation
energies and corresponding eigenstates of reference model Hamiltonian respectively. Using
equation (1), we evaluated exchange constants for spin rings and all-to-all systems having
S = 1/2. Half-filled Hubbard model has been used as reference for exchange constants
calculation. We compared eigenvalues spectra of both models in order to assess quality and
applicability of proposed method (Fig. 1). The results have also been checked with existing
approaches [1, 2] for evaluation of exchange constants.
Fig. 1 Nearest neighbor and next nearest neighbor exchange interactions of spin ring evaluated
by (1) for different parameters t and U of Hubbard model (a) and corresponding energy
excitation spectra (b).
The work is supported by the grant program of the Russian Science Foundation 15-12-20021.
[1] Anderson PW. Phys. Rev. 1959; 115:2.
[2] Liechtenstein AI, Katsnelson MI, et al. J. Magn. Magn. Mater. 1987; 67:65
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–53–
THE ELECTRONIC STRUCTURE OF A MONOLAYER FeSe ON THE
SrTiO3 SUBSTRATE
I.A. Nekrasova, N.S. Pavlova, M.V. Sadovskiia,b, A.A. Slobodchikova*
aInstitute for Electrophysics, Russian Academy of Sciences, Ural Branch, Amundsen Str. 106,
Ekaterinburg, 620016, Russia
bM.N. Mikheev Institute for Metal Physics, Russian Academy of Sciences,
Ural Branch, S. Kovalevsky Str. 18, Ekaterinburg, 620290, Russia
The discovery of a new class of superconductors based on iron pnictides has opened up the
new prospects for the study of high-temperature superconductivity (cf. Reviews [1]). A
significant breakthrough in the study of iron based superconductors happened with the
observation of a record high Tc (higher than 50K) in epitaxial films of single FeSe monolayer on
a substrate of SrTiO3 (STO) [2].
To calculate electronic properties of the FeSe monolayer film on SrTiO3 substrate we used
the Quantum-Espresso package [3]. By looking on left panel of Fig.1 one can immediately
recognize that there appears an additional hole band near the M-point. To understand its origin
we plotted on right panel of Fig.1 O-2p states of topmost surface TiO2 layer of STO substrate.
We can conclude [4], that the presence of STO interface leads to the appearance of this
additional band of O-2p surface states near the Fermi level. Also we observe rather small
splitting of electron bands at M-point, which may have the relation to the observation of the
«replica» band at M-point [5], in contrast to optical phonons in STO scenario.
Fig. 1 (left) — LDA calculated band dispersion of FeSe monolaer on SrTiO3 substrate;
(right) — LDA calculated O-2p states of surface TiO2 layer of STO substrate.
Sadovskii M V. Usp. Fiz. Nauk 178 1243 (2008)
Wang Qing-Yan et al., Chin. Phys. Lett. 29 037402 (2012)
Giannozzi Paolo et al., J. Phys.: Condens. Matter 21 395502 (2009)
I.A. Nekrasov et al., arXiv:1605.02404
Lee J J et al., Nature 515 245 (2014)
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–54–
ELECTRONIC STRUCTURE AND MAGNETIC PROPERTIES OF
HEXAGONAL AND CUBIC MODIFICATIONS OF ALUMINIUM
NITRIDE DOPED WITH SP-IMPURITIES (B, C, O)
V.V. Bannikov 1*, V.S. Kudyakova 2, A.A. Elagin2, M.V. Baranov 2, A.R. Beketov 2
1 Institute of Solid State Chemistry, Ural Branch RAS, Russia
2 Ural Federal University, Russia
The comparative modeling of band structure and magnetic properties of aluminium nitride
hexagonal and cubic modifications (w-AlN and r-AlN, respectively) with sp-impurities
(X=B,C,O) introduced into anionic sub-lattice (see Fig.1) has been performed by ab initio
FLAPW-GGA method. It was shown that the implantation of B or C into w-AlN results in spin
polarization of its near-Fermi electronic states, which is close to 100% (see Fig.2), as well, as in
considerable magnetic moments (~0.3–0.4 B) induced on the impurity atoms, so w-
AlN0.75(B,C)0.25 systems may be characterized as magnetic half-metals and so-called d0-
magnetics (see J.M.D. Coey, Sol. St. Sci., 7, 660 (2005)). However, this kind of situation does
not take place for cubic r-AlN0.75X0.25 systems predicted to be usual non-magnetic metals (see
Fig.2). The estimated substitution energies of X dopants for nitrogen in original structure are
~1.2-1.5 times higher for w-AlN0.75(B,C)0.25 systems as compared with r-AlN0.75(B,C)0.25,
indicating that the formation of the latter ones is expected to be more preferable. From the other
hand, the energy of oxygen substitution for r-AlN is ~1.5 times higher than for w-AlN, so the
metastable r-AlN phase is expected to be more resistant to oxidation processes than w-AlN is.
Fig. 1 The extended cells of
r-AlN0.75X0.25 (upper) and w-
AlN0.75X0.25 (lower).
Fig. 2 Spin-polarized densities of electronic states calculated for
w-AlN0.75X0.25 (upper row) and r-AlN0.75X0.25 (lower row). a:
X=B; b: X=C.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–55–
CALCULATION SCHEME BASED ON THE EXTENDED EQUATIONS
OF DMFT FOR SQUARE AND TRIANGULAR LATTICES
Daria Medvedeva 1*, V.V. Mazurenko1, S.N. Iskakov1,2, A.I. Lichtenstein1,3
1 Ural Federal University, Russia
2 Department of Physics, University of Michigan, USA
3 Hamburg University, Germany
DMFT is successful route for description of strongly correlated systems, which can exhibit
some of the most intriguing features known to condensed matter physics, including high-
temperature superconductivity, heavy fermion behavior, metal-insulator transitions and others
[1]. However, there are many examples when one should overcome the main limitation of the
DMFT that is neglect of the non-local magnetic and charge correlations. This is the case for
graphene-based systems for which strong inter-site Coulomb correlations [2] and magnetic
couplings [3] were found. To solve this problem one can use different extensions and
modifications of the single-site DMFT approach that are cluster DMFT[4], dual fermions
approach[5] and others.
In this work we propose a distinct numerical scheme based on the exact diagonalization
approach to solve the equations of the extended dynamical mean-field theory. In contrast to the
single-site DMFT we deal with the impurity problem where the correlated site interacts both
with fermion and boson reservoirs.
As a result, one can see that the account of the non-local magnetic interactions, leads to
the formation or gain of the low-energy peaks of the local magnetic susceptibility.
[1] A. Georges, G. Kotliar, W. Krauth and M.J. Rosenberg, Review Modern Physics, 68 , 13
(1996).
[2] T. O. Wehling, E. Sasioglu, C. Friedrich, A. I. Lichtenstein, M. I. Katsnelson, and S. Blugel,
Phys. Rev. Lett. 106, 236805 (2011).
[3] A. N. Rudenko, F. J. Keil, M. I. Katsnelson, and A. I. Lichtenstein, Phys. Rev. B 88 ,
081405 (2013).
[4] H. Park, K. Haule and G. Kotliar, Phys. Rev. Let. 101 , 186403 (2008).
[5] S. Brener, H. Hafermann, A.N. Rubtsov, M.I. Katsnelson and A.I. Lichtenstein, Physical
Review B, 77 , 195105 (2008).
*Email: [email protected]
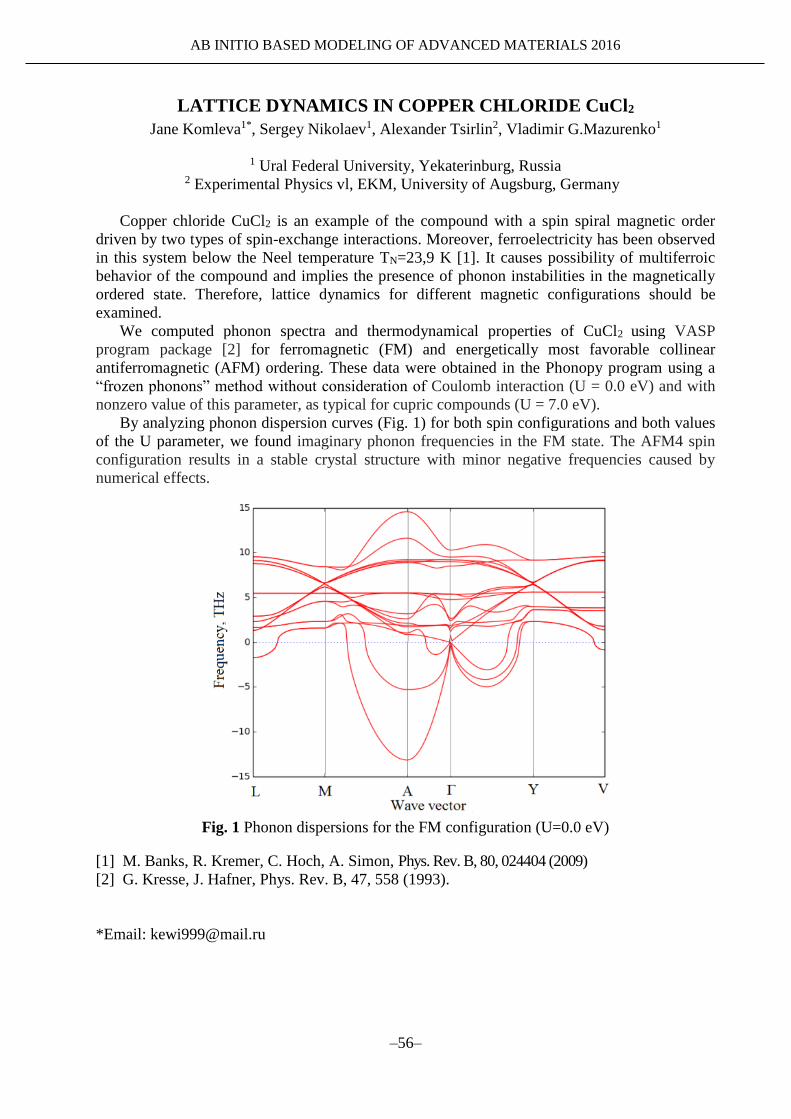
AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–56–
LATTICE DYNAMICS IN COPPER CHLORIDE CuCl2
Jane Komleva1*, Sergey Nikolaev1, Alexander Tsirlin2, Vladimir G.Mazurenko1
1 Ural Federal University, Yekaterinburg, Russia
2 Experimental Physics vl, EKM, University of Augsburg, Germany
Copper chloride CuCl2 is an example of the compound with a spin spiral magnetic order
driven by two types of spin-exchange interactions. Moreover, ferroelectricity has been observed
in this system below the Neel temperature TN=23,9 K [1]. It causes possibility of multiferroic
behavior of the compound and implies the presence of phonon instabilities in the magnetically
ordered state. Therefore, lattice dynamics for different magnetic configurations should be
examined.
We computed phonon spectra and thermodynamical properties of CuCl2 using VASP
program package [2] for ferromagnetic (FM) and energetically most favorable collinear
antiferromagnetic (AFM) ordering. These data were obtained in the Phonopy program using a
“frozen phonons” method without consideration of Coulomb interaction (U = 0.0 eV) and with
nonzero value of this parameter, as typical for cupric compounds (U = 7.0 eV).
By analyzing phonon dispersion curves (Fig. 1) for both spin configurations and both values
of the U parameter, we found imaginary phonon frequencies in the FM state. The AFM4 spin
configuration results in a stable crystal structure with minor negative frequencies caused by
numerical effects.
Fig. 1 Phonon dispersions for the FM configuration (U=0.0 eV)
[1] M. Banks, R. Kremer, C. Hoch, A. Simon, Phys. Rev. B, 80, 024404 (2009)
[2] G. Kresse, J. Hafner, Phys. Rev. B, 47, 558 (1993).
*Email: [email protected]
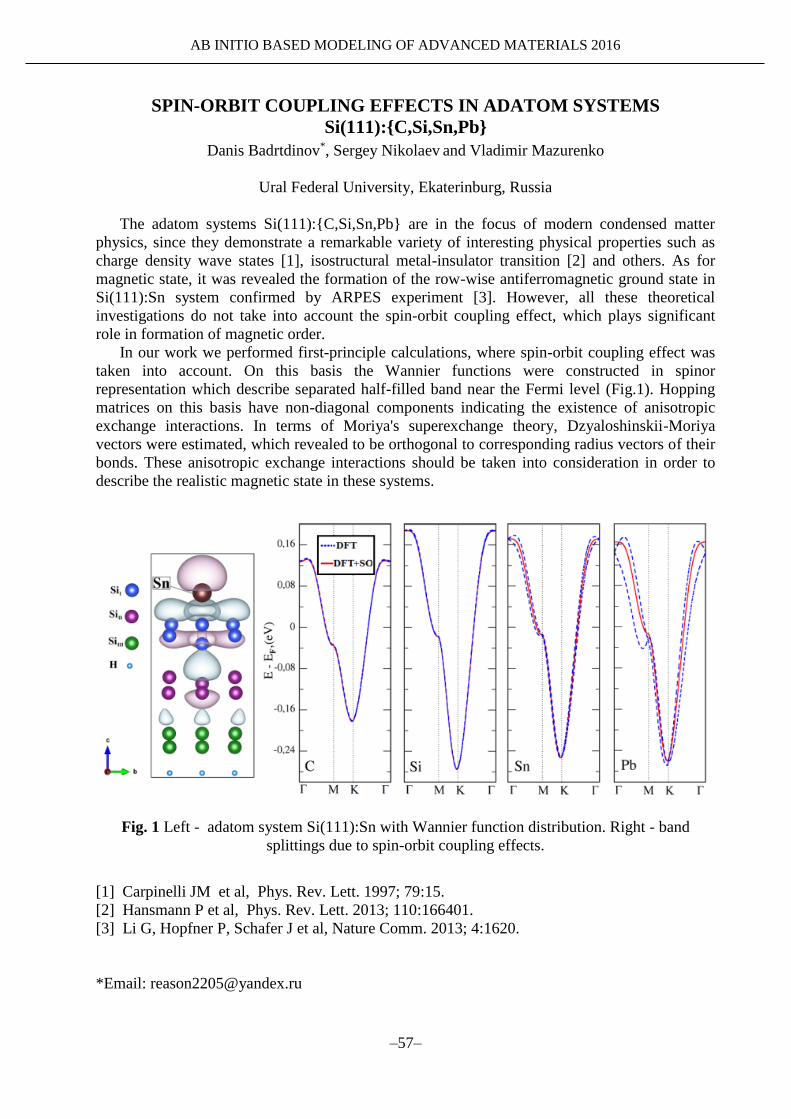
AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–57–
SPIN-ORBIT COUPLING EFFECTS IN ADATOM SYSTEMS
Si(111):{C,Si,Sn,Pb}
Danis Badrtdinov*, Sergey Nikolaev and Vladimir Mazurenko
Ural Federal University, Ekaterinburg, Russia
The adatom systems Si(111):{C,Si,Sn,Pb} are in the focus of modern condensed matter
physics, since they demonstrate a remarkable variety of interesting physical properties such as
charge density wave states [1], isostructural metal-insulator transition [2] and others. As for
magnetic state, it was revealed the formation of the row-wise antiferromagnetic ground state in
Si(111):Sn system confirmed by ARPES experiment [3]. However, all these theoretical
investigations do not take into account the spin-orbit coupling effect, which plays significant
role in formation of magnetic order.
In our work we performed first-principle calculations, where spin-orbit coupling effect was
taken into account. On this basis the Wannier functions were constructed in spinor
representation which describe separated half-filled band near the Fermi level (Fig.1). Hopping
matrices on this basis have non-diagonal components indicating the existence of anisotropic
exchange interactions. In terms of Moriya's superexchange theory, Dzyaloshinskii-Moriya
vectors were estimated, which revealed to be orthogonal to corresponding radius vectors of their
bonds. These anisotropic exchange interactions should be taken into consideration in order to
describe the realistic magnetic state in these systems.
Fig. 1 Left - adatom system Si(111):Sn with Wannier function distribution. Right - band
splittings due to spin-orbit coupling effects.
[1] Carpinelli JM et al, Phys. Rev. Lett. 1997; 79:15.
[2] Hansmann P et al, Phys. Rev. Lett. 2013; 110:166401.
[3] Li G, Hopfner P, Schafer J et al, Nature Comm. 2013; 4:1620.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–58–
LEAD OXYHALIDES Pb3X2O2 (X=Cl, Br, I): AB INITIO CALCULATIONS
OF PHONON SPECTRA AND OPTICAL PROPERTIES
Dmitry Zakir’yanov 1*, Vladimir Chernyshev 1
1 Ural Federal University, Ekaterinburg, Russia
Lead oxyhalides Pb3X2O2 (X=Cl, Br, I) are interesting as a materials with scpecific optic and
ion-cunduction properties [1–3].
Ab initio calculations of the crystal structure and the phonon spectrum of Pb3O2X2 (X=Cl,
Br, I) were performed in the framework of the density functional theory using PBE0 [4]
functional. The CRYSTAL code, designed for periodic systems, was used. A good agreement
with the experimental data has been reached [5]. The results of calculations have made it
possible to interpret the experimental vibration spectra and reveal silent modes, which do not
manifest themselves in these spectra but influence the optical properties of the crystal.
Interpretation of phonon spectra was performed.
Components of the dielectric tensor, birefringence, angle between optic axes and range of
thermodynamic parameters were calculated. In particular, the calculations results shown that the
anisotropy of Pb3O2Br2 will be less pronounced than of Pb3O2Cl2.
Changes in the physical properties of crystals in a sequence Pb3O2Cl2 → Pb3O2Br2 →
Pb3O2I2 were studied by ab initio calculations.
This work was supported by the Russian Foundation for Basic Research, project no. 15-03-
00368a.
[1] Sigman MB, Korgel BA. J. Am. Chem. Soc. 2005; 127:10089.
[2] Siidra OI, Krivovichev SV, Depmeier W. Phys. Chem. 2007; 414:501.
[3] Withers NJ, Akins BA, Rivera AC, Plumley JB, Smolyakov GA, Osinski M. SPIE
Proceedings 2009; 73041N:1.
[4] Perdew JP, Ernzerhof M, Burke K.J. Chem. Phys. 1996; 105:9982.
[5] Zakir’yanov DO, Chernyshev VA, Zakir’yanova ID. Physics of the Solid State 2016;
58:325.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–59–
THE KOHN ANOMALIES IN THREE-DIMENSIONAL SYSTEMS
A. Stepanenko1*, D. Vesnina1, P. Igoshev2,1, A. Katanin2,1
1Ural Federal University, 620002, Ekaterinburg, Russia
2Institute of Metal Physics, 620990, Ekaterinburg, Russia
The model approaches to itinerant systems allow explaining experimental data and predicting
properties of new materials. These approaches highlight essential peculiarities of the electronic
spectrum, such as van Hove singularities [1] and Kohn anomalies [2,3], which play important
role in physical properties of itinerant systems. While the van Hove singularities lead to
ferromagnetism, Kohn anomalies are important for systems with the spin density wave order.
In this study we investigate a possibility of Kohn anomalies, which are accompanied by a
maximum of the non-uniform magnetic susceptibility at the wave vector 2kF, in systems with
face centered cubic (fcc) and body-centered cubic (bcc) lattice. While the fcc lattice yields such
Kohn anomalies already within the one band model with nearest-neighbor hopping, the bcc
lattice does not possess this property. At the same time, including more distant neighbors and/or
several (at least two) bands yields Kohn anomalies within relatively simple itinerant models on
bcc lattice.
As an example, we consider the model of the two relevant bands of chromium including
hopping between first, second and third neighbors. The fit of the Fermi surfaces was used to
determine Kohn points and study the effect of magnetic correlations within the model approach.
The momentum dependence of magnetic susceptibility at the the non-zero and zero temperature
in the paramagnetic phase is calculated.
Fig. 1 The Fermi surfaces of Cr in the two band model.
[1] van Hove L. Phys. Rev. 1953; 89: 1189.
[2] Kohn W. Phys. Rev. Lett. 1959; 2:393; Rice TM. Phys. Rev. B 1970; 2:9.
[3] Schäfer T, Katanin AA, Held K, Toschi A. arXiv:1605.06355.
*Email: [email protected]
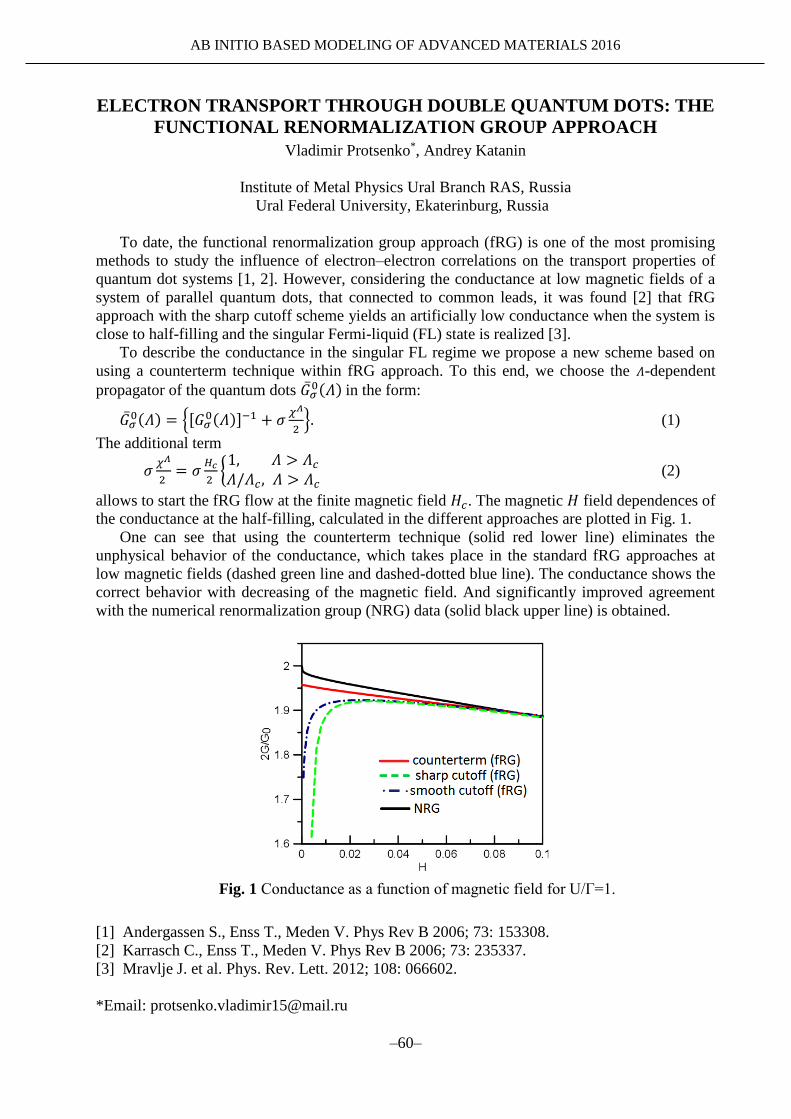
AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–60–
ELECTRON TRANSPORT THROUGH DOUBLE QUANTUM DOTS: THE
FUNCTIONAL RENORMALIZATION GROUP APPROACH
Vladimir Protsenko*, Andrey Katanin
Institute of Metal Physics Ural Branch RAS, Russia
Ural Federal University, Ekaterinburg, Russia
To date, the functional renormalization group approach (fRG) is one of the most promising
methods to study the influence of electron–electron correlations on the transport properties of
quantum dot systems [1, 2]. However, considering the conductance at low magnetic fields of a
system of parallel quantum dots, that connected to common leads, it was found [2] that fRG
approach with the sharp cutoff scheme yields an artificially low conductance when the system is
close to half-filling and the singular Fermi-liquid (FL) state is realized [3].
To describe the conductance in the singular FL regime we propose a new scheme based on
using a counterterm technique within fRG approach. To this end, we choose the 𝛬-dependent
propagator of the quantum dots �̅�𝜎0(𝛬) in the form:
�̅�𝜎0(𝛬) = {[𝐺𝜎
0(𝛬)]−1 + 𝜎𝜒𝛬
2}. (1)
The additional term
𝜎𝜒𝛬
2 = 𝜎
𝐻𝑐
2 {
1, 𝛬 > 𝛬𝑐
𝛬/𝛬𝑐 , 𝛬 > 𝛬𝑐 (2)
allows to start the fRG flow at the finite magnetic field 𝐻𝑐. The magnetic 𝐻 field dependences of
the conductance at the half-filling, calculated in the different approaches are plotted in Fig. 1.
One can see that using the counterterm technique (solid red lower line) eliminates the
unphysical behavior of the conductance, which takes place in the standard fRG approaches at
low magnetic fields (dashed green line and dashed-dotted blue line). The conductance shows the
correct behavior with decreasing of the magnetic field. And significantly improved agreement
with the numerical renormalization group (NRG) data (solid black upper line) is obtained.
Fig. 1 Conductance as a function of magnetic field for U/Γ=1.
[1] Andergassen S., Enss T., Meden V. Phys Rev B 2006; 73: 153308.
[2] Karrasch C., Enss T., Meden V. Phys Rev B 2006; 73: 235337.
[3] Mravlje J. et al. Phys. Rev. Lett. 2012; 108: 066602.
*Email: [email protected]
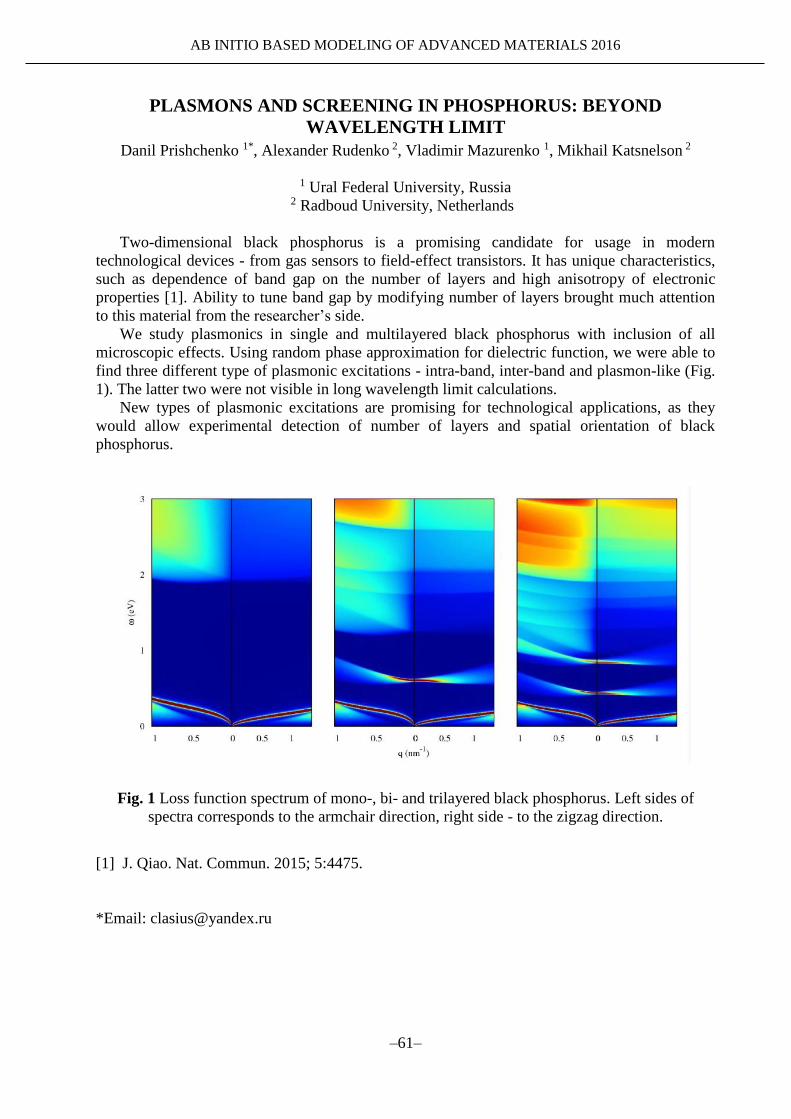
AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–61–
PLASMONS AND SCREENING IN PHOSPHORUS: BEYOND
WAVELENGTH LIMIT
Danil Prishchenko 1*, Alexander Rudenko 2, Vladimir Mazurenko 1, Mikhail Katsnelson 2
1 Ural Federal University, Russia
2 Radboud University, Netherlands
Two-dimensional black phosphorus is a promising candidate for usage in modern
technological devices - from gas sensors to field-effect transistors. It has unique characteristics,
such as dependence of band gap on the number of layers and high anisotropy of electronic
properties [1]. Ability to tune band gap by modifying number of layers brought much attention
to this material from the researcher’s side.
We study plasmonics in single and multilayered black phosphorus with inclusion of all
microscopic effects. Using random phase approximation for dielectric function, we were able to
find three different type of plasmonic excitations - intra-band, inter-band and plasmon-like (Fig.
1). The latter two were not visible in long wavelength limit calculations.
New types of plasmonic excitations are promising for technological applications, as they
would allow experimental detection of number of layers and spatial orientation of black
phosphorus.
Fig. 1 Loss function spectrum of mono-, bi- and trilayered black phosphorus. Left sides of
spectra corresponds to the armchair direction, right side - to the zigzag direction.
[1] J. Qiao. Nat. Commun. 2015; 5:4475.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–62–
CONTACTS OF CARBON AND GOLD NANOTUBES:
FIRST PRINCIPLES CALCULATIONS
Sergey Sozykin 1*, Valery Beskachko 1
1 South Ural State University, Chelyabinsk, Russia
Expectations to create a new generation of electronic devices having very small dimensions,
associated with carbon nanotubes (CNT). One of the potential problems that prevent the
implementation of such devices is the high resistance of the CNT-metal contact. Experimental
studies of a number of metals for ohmic contacts lead to contradictory conclusions because of
the difficulty of experimental conditions reproduction. It is thought that the most promising
materials for the contacts are Pd, Ti, Au and Pt. There are also theoretical studies of CNT-metal
contacts. However, they rarely relate to the current-voltage characteristics. In particular, these
characteristics have not been studied for the «end» contact geometry where metal electrodes are
attached to the ends of the tube. Doped metal tube edges are often considered as electrodes.
By now nanotubes obtained not only from carbon, but also from many other elements and
compounds, in particular from noble metals, including the mentioned above (Au, Pt, Pd). Unlike
carbon nanotubes, all metal tubes are conductors and often regarded as wires in nanodevices. It
is interesting to consider how such tubes will act as «end» electrodes. The answer to this
question we have tried to obtain by means of computer simulation.
It is convenient for simulation that the diameters of the CNT and noble metal tubes can be
chosen close. Thus, a CNT (7,7) diameter is close to the diameter of a gold nanotube (10,0). In
our calculations we used models of these nanotubes of 112 and 40 atoms, respectively. The
simulation was performed using the density functional theory and non-equilibrium Green's
functions as implemented in the SIESTA package. We used the exchange-correlation functional
PBE and DZP basis set.
We have determined the equilibrium geometry of the system and electrical charges on atoms
near the C-Au boundary, mechanical strength of the contact and the current-voltage
characteristic of the system.
a b c d
Fig. 1 Atomic structure of (7,7) carbon (a,b) and (10,0) gold (c,d) nanotubes
*Email: [email protected]
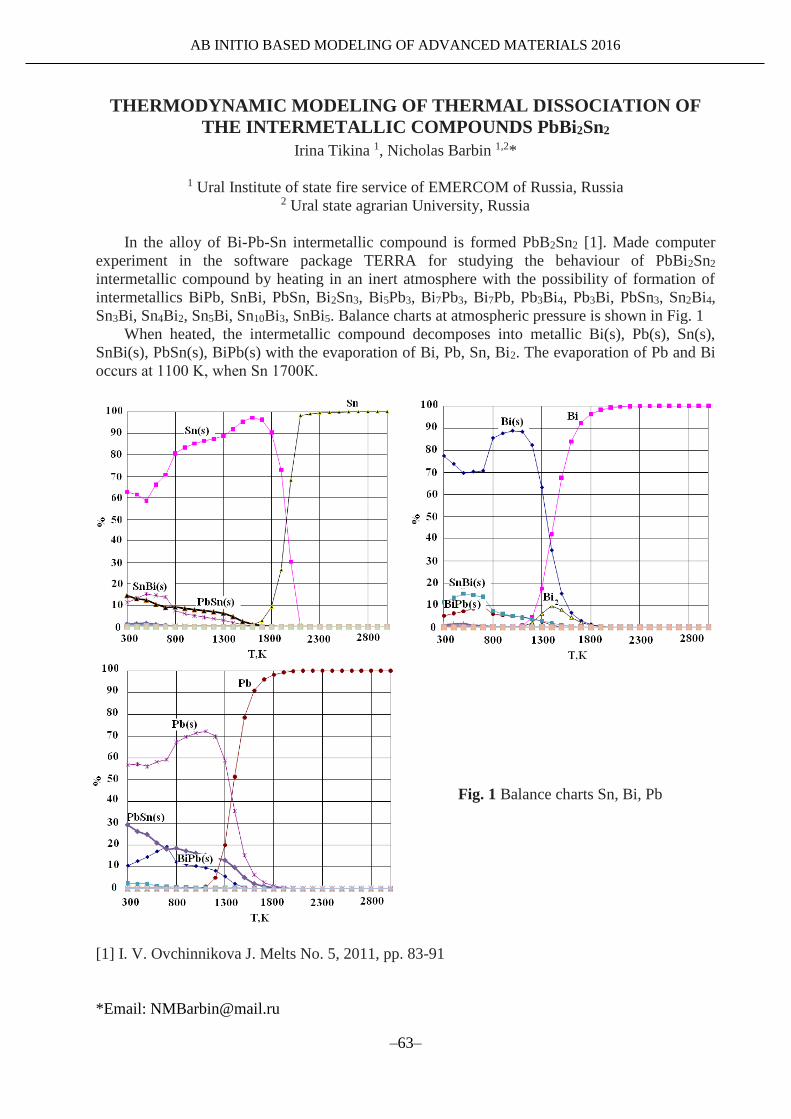
AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–63–
THERMODYNAMIC MODELING OF THERMAL DISSOCIATION OF
THE INTERMETALLIC COMPOUNDS PbBi2Sn2
Irina Tikina 1, Nicholas Barbin 1,2*
1 Ural Institute of state fire service of EMERCOM of Russia, Russia
2 Ural state agrarian University, Russia
In the alloy of Bi-Pb-Sn intermetallic compound is formed PbB2Sn2 [1]. Made computer
experiment in the software package TERRA for studying the behaviour of PbBi2Sn2
intermetallic compound by heating in an inert atmosphere with the possibility of formation of
intermetallics BiPb, SnBi, PbSn, Bi2Sn3, Bi5Pb3, Bi7Pb3, Bi7Pb, Pb3Bi4, Pb3Bi, PbSn3, Sn2Bi4,
Sn3Bi, Sn4Bi2, Sn5Bi, Sn10Bi3, SnBi5. Balance charts at atmospheric pressure is shown in Fig. 1
When heated, the intermetallic compound decomposes into metallic Bi(s), Pb(s), Sn(s),
SnBi(s), PbSn(s), BiPb(s) with the evaporation of Bi, Pb, Sn, Bi2. The evaporation of Pb and Bi
occurs at 1100 K, when Sn 1700К.
Fig. 1 Balance charts Sn, Bi, Pb
[1] I. V. Ovchinnikova J. Melts No. 5, 2011, pp. 83-91
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–64–
MAGNETIC ANISOTOPY EFFECTS IN FE-GA ALLOYS
Petrik Mikhail 1,2*, Danis Badrtdinov 3
1 Institute of Metal Physics Ural Branch RAS, Russia
2Institute of quantum materials science, Russia 3 Ural Federal University, Russia
The Fe-Ga alloy attract significant attention since of its abnormally high magnetostriction
[1]. At concentrations of Ga below 19% it appears due to the effect of single Ga atoms in Fe
matrix [2]; at higher concentrations the short-range order plays important role [3]. In order to
obtain the realistic picture of this phenomena on the microscopic level and estimate
magnetostriction by the formula:
)2
/2(3
)/(2
100
dtot
Ed
dMCA
dE
,
(1)
the calculation of the magnetocrystalline anisotropy energy EMCA is necessary. On the basis of
ab initio calculations performed using VASP, the electronic structure of Fe-Ga alloy was
obtained and localized Wannier functions were constructed. These Wannier functions provide
accurate description of the energy spectrum of the system in a wide range of energies. The
Green's functions were constructed from hopping matrices in the basis of Wannier functions
and, considering the weak spin-orbital coupling in this system, the perturbation theory methods
were utilized for the estimation of magnetocrystalline anisotropy energy [4]. This approach can
be considered as very useful for describing magnetoelastic properties of systems with small
MAEs.
A.E. Clark, J.B. Restorff, M. Wun-Fogle , T.A. Lograsso, D.L. Schlagel // IEEE Trans.
Magn. — 2000. —Vol. 36, No. 5 — P. 3238–3240
R. Wu // Journal of Applied Physics. — 2002. — Vol. 91. — P. 7358.
M.V.Petrik, O.I.Gorbatov, Yu.N.Gornostyrev // JETP Letters. — 2014. — V. 98. — P.
809—812.
I. V. Solovyev, P. H. Dederichs, I. Mertig // Physical Review B. – 2011. — Vol. 52, No. 18.
— P. 13419.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–65–
STRUCTURE AND LATTICE DYNAMICS OF BiMnO3: AB INITIO
CALCULATIONS
D. Nazipov1*, A. Nikiforov1, L. Gonchar1,2
1Ural Federal University, Ekaterinburg, Russia 2Ural State University of Railway Transport, Ekaterinburg, Russia
Multiferroics are actively studied and have a wide practical application. The effect of
ferroelectricity and at the same time long-range magnetic order in multiferroic opens the
possibility of creation of electronic devices with controllable magnetic characteristics. At the
present time being searched for multiferoics combining magnetic and ferroelectric properties.
Among the materials, perovskite-like BiMnO3 has attracted particular attention of researchers.
Complicated orbital structure [1,2] on Mn3+ ions leads to ferromagnetic ordering below
Tc=100K and existence of lone pair of 6s2 electrons of Bi3+ may lead to ferroelectric effect.
Thus, a detailed analysis of the microscopic mechanisms of interaction between the magnetic
and crystal subsystems is required.
In this paper we present the results of ab initio calculations of the crystal structure
parameters and the phonon spectrum of BiMnO3. Lattice constants, coordinates of ions in cell
and bond lengths have been obtained for the ferromagnetic C2/с phase in a good agreement with
experiment [3]. Calculated frequencies and intensities of the active modes in IR and Raman
spectra have been compared with the experiment [4].
Calculations have been performed in solid state calculations package CRYSTAL14 [5] in the
framework of MO LCAO approximation, using density functional theory method with hybrid
DFT/HF functionals. The ions are described by full-electron Gauss-type basis sets and
pseudopotential for Bi3+.
In model approach orbital ordering and splitting of 5D (3d4) term of two nonequivalent Mn3+
ions have been obtained using crystal field approximation.
A.M. dos Santos, A.K. Cheetham, T. Atou, Y. Syono, Y. Yamaguchi, K. Ohoyama, H.
Chiba, C.N.R. Rao. Phys. Rev. B 66, 064425 (2002).
L.E. Gonchar, A.E. Nikiforov. Phys. Rev. B 88, 094401 (2013).
A.A. Belik, S. Likubo, T. Yokosawa, K. Kodama, N. Igawa, S. Shamoto, M. Azuma, M.
Takano, K. Kimoto, Y. Matsui, E. Takayama-Muromachi. J. Am. Chem. Soc. 129, 971
(2007).
W. S. Mohamed, A. Nucara, G. Calestani, F. Mezzadri, E. Gilioli, F. Capitani, P. Postorino,
P. Calvani. Phys. Rev. B 92, 054306 (2015).
URL: http://www.crystal.unito.it/index.php
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–66–
STRUCTURE AND LATTICE DYNAMICS OF PrFe3(BO3)4:
AB INITIO CALCULATION
Vladimir Chernyshev1*, Anatoliy Nikiforov 1, Vladislav Petrov 1
1 Ural Federal University, Russia
The crystal structure and phonon spectrum of PrFe3(BO3)4 are ab initio calculated in the
context of the density functional theory [1,2]. The ion coordinates in the unit cell of a crystal and
the lattice parameters are evaluated from the calculations. The types and frequencies of the
fundamental vibrations, as well as the line intensities of the IR spectrum, are determined. The
elastic constants of the crystal are calculated. A “seed” frequency of the vibration strongly
interacting with the electron excitation on the praseodymium ion is obtained for low-frequency
A2 mode. The calculated results are in agreement with the known experimental data.
[1] Tsirel’son V. G., Quantum Chemistry: Molecules, Molecular Systems and Solids, Moscow:
Binom; 2010 [in Russian].
[2] Dovesi R., Saunders V. R., Roetti C., Orlando R., Zicovich-Wilson C. M., Pascale F.,
Civalleri B., Doll K., Harrison N. M., Bush I. J., D’Arco Ph., Llunell M., CRYSTAL09
User’s Manual (University of Torino, Torino, Italy, 2009).
http://www.crystal.unito.it/index.php, 2009.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–67–
LANTHANIDE-DOPED GERMANATES: DFT STUDY OF LATTICE
DYNAMICS AND ELECTRONIC STRUCTURE OF OPTICAL HOSTS
Ivan Leonidov 1*, Vladislav Petrov 2, Vladimir Chernyshev 2,
Alexey Ishchenko 2, Ekaterina Konstantinova 1, Anatoliy Nikiforov 2
1 Institute of Solid State Chemistry Ural Branch RAS, Russia
2 Ural Federal University, Russia
Multifunctional materials based on the M2Ge7O16 (M = Ca, Cd), CaY2Ge4O12, CaLa2Ge3O10
germanates are of considerable interest for their applications in solid state electrochemistry and
optical engineering [1, 2]. While these compounds doped with lanthanide ions have been
extensively studied by different experimental techniques, DFT-aided interpretation of their
crystal structures and physical/chemical properties has been discussed in few recent papers [3,
4].
A series of DFT functionals has been applied in describing the equilibrium crystal structure,
FTIR and Raman spectra, and electronic properties of Ca2Ge7O16, CaY2Ge4O12, CaLa2Ge3O10.
Calculations have been performed with the periodic ab initio CRYSTAL code using all electron
Gaussian-type basis sets. The following exchange-correlation functionals have been tested: LDA
functional (SVWN), semilocal GGA tailored for molecules (PBE), the functionals devised for
solids (PBEsol, WC), and several hybrids (B3LYP, WC1LYP, PBE0, PBE(n = 6)). Whereas
GGA functionals reproduce quite well the crystal structure, they fail for the phonon spectra and
electronic structure of the studied oxides. The inclusion of HF exchange in the hybrid
functionals leads to very good predicted structures of the germanates of different types. The
most accurate geometry description and optical band gap estimation, and the best agreement
between the recorded infrared and Raman spectra and their computed counterparts have obtained
with the WC1LYP and PBE (n = 6) hybrid functionals.
This work was partially supported by the RFBR Grant No. 14–03–31324. Computational
procedures were carried out at the URAN Computing Platform at IMM UB RAS (Ekaterinburg,
Russia). I. L. would like to acknowledge support from the Russian President Fellowship SP–
931.2016.1.
Aravindan V, Lee YS, Madhavi S. Adv Energy Mater 2015; 5:1402225.
Leonidov II, Zubkov VG, Tyutyunnik AP, Tarakina NV, Surat LL, Koryakova OV,
Vovkotrub EG. J Alloys Compd 2011; 509:1339.
Leonidov II, Petrov VP, Chernyshev VA, Nikiforov AE, Vovkotrub EG, Tyutyunnik AP,
Zubkov VG. J Phys Chem C 2014; 118:8090.
Wang T, Gou J, Xu XH, Zhou DC, Qiu JB, Yu X. Opt Express 2015; 23:12595.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–68–
ELECTRONIC STRUCTURE AND MAGNETIC PROPERTIES OF THE
STRONG-RUNG SPIN-1 LADDER Rb3Ni2(NO3)7
ZlataV. Pchelkina1,2*, O. S. Volkova2,3, V. V. Mazurenko2, A. N. Vasiliev2,3,4
1 M.N. Miheev Institute of Metal Physics of Ural Branch of RAS, Ekaterinburg, Russia 2Department of Theoretical Physics and Applied Mathematics, Ural Federal University,
Ekaterinburg, Russia 3 Lomonosov Moscow State University, Moscow, Russia
4National University of Science and Technology “MISiS,” Moscow, Russia
The electronic structure and magnetic properties of Rb3Ni2(NO3)7, the prototype material of
the strong-rung spin-1 ladder model, was calculated within LDA+U approximation. The
isotropic exchange, Dzyaloshinskii-Moriya interactions and single-ion anisotropy tensor were
calculated by using the magnetic force theorem with different types of perturbations [1, 2]. The
analysis of partial orbital contributions into the matrix of exchange integrals reveals the reason
of strong exchange along the rungs of the ladder. The Heisenberg model with obtained
interatomic exchange interactions was solved by quantum Monte Carlo method. The model
magnetization and spin susceptibility, as well as theoretical estimation of the Curie-Weiss
temperature are in good agreement with the experimental one. The several plateaus in the filed
dependence of magnetization at low temperatures are predicted.
[1] Lichtenstein A I, Katsnelson M I, Antropov V P, and Gubanov V A, J. Magn Magn Mater
1987; 67:65.
[2] Mazurenko V V and Anisimov V I, Phys Rev B 2005; 71:184434.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–69–
FULL POTENTIAL STUDY OF ELECTRONIC AND MAGNETIC
PROPERTIES OF FUNCTIONALISED GRAPHENE.
Ilya Piterskikh 1*, Danil Boukhvalov 1,2, Vladimir Mazurenko 1
1 Institute of Physics and Technology, Ural Federal University, Ekaterinburg, Russia
2 Department of Chemistry, Hanyang University, Seoul, Republic of Korea
Numerous studies have shown high sensitivity of graphene electron and its energy structure
to defects of various kinds, including the atoms adsorbed on its surface, thus confirming that the
chemical modification of graphene is a promising direction of the management of its electronic
structure. Due to the fact that a significant portion of the above studies made on the basis of the
pseudopotential method, it is of interest to study the changes in the electronic structure of
graphene with the density functional theory (DFT) on the basis of the full-potential wave
function using linearized augmented plane wave method (FP-LAPW). Research was conducted
with Wien2k package, exchange and correlation energies were evaluated with the local density
approximation (LDA).
The results of changes in the electronic structure and magnetic properties of graphene at
various numbers of adsorbed atoms of hydrogen and fluorine were presented. It is found that the
magnetic properties of graphane and fluorographene depend on the distance between adjacent
atoms adsorbed, and the distance between the fluorine atom and its associated carbon atom in
the lattice of graphene. The features of the band structure and the electron density as a function
of fluorine concentration and the degree of deformation of the graphene planes during the
adsorption of fluorine were studied. The effect of the concentration of adsorbed fluorine as a
factor of mutual influence of interaction between neighboring fluorine atoms on the electron
graphene structure (C2F, C4F and C18F) was shown.
It is shown that the magnetic properties of graphane and fluorographene depend on the
distance between adjacent atoms adsorbed, and the distance between the fluorine atom and its
associated carbon atom in the graphene lattice.
[1] http://www.wien2k.at/
[2] Rudenko A.N. et al. Phys. Rev. B. 88, 081405 (2013)
[3] Fabian J. et al. Physical Review B (PRB) 91, 115141 (2015)
[4] Boukhvalov D. et al. J Phys Cond Matter 2009; 21(34):344205.Manning JR. Diffusion
kinetics for atoms in crystals, London: Van Nostrand; 1968.
*Email: [email protected]
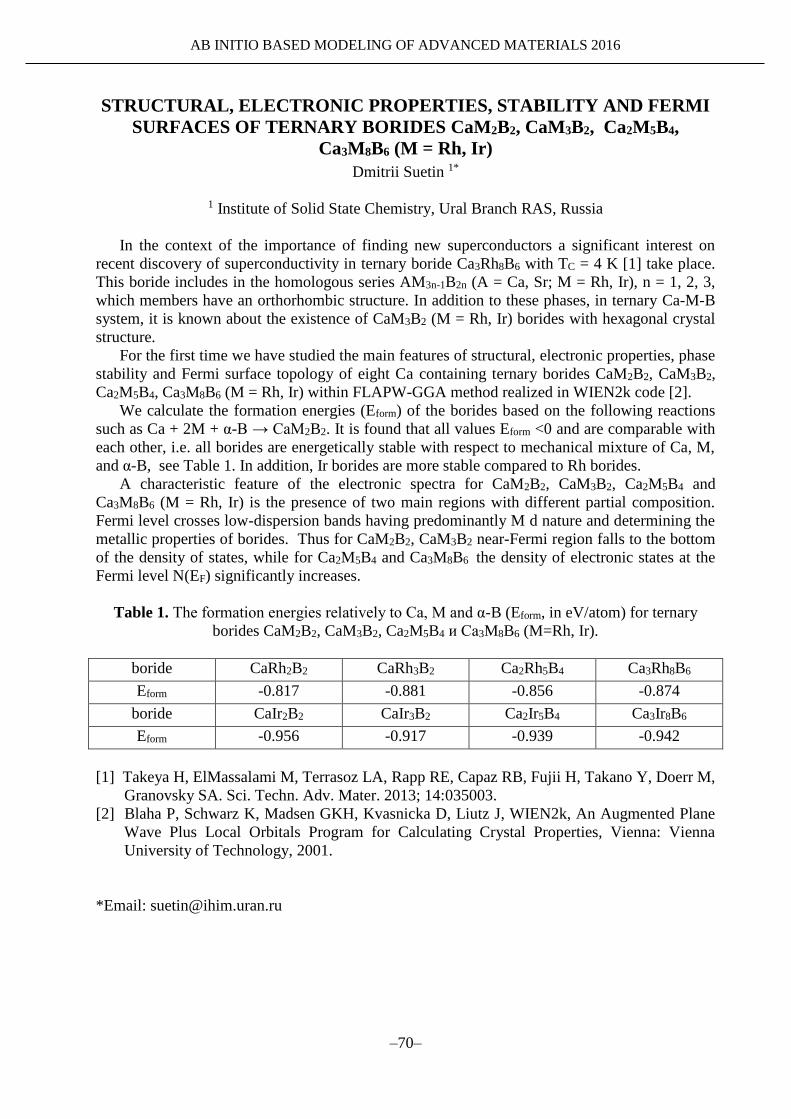
AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–70–
STRUCTURAL, ELECTRONIC PROPERTIES, STABILITY AND FERMI
SURFACES OF TERNARY BORIDES CaM2B2, CaM3B2, Ca2M5B4,
Ca3M8B6 (M = Rh, Ir)
Dmitrii Suetin 1*
1 Institute of Solid State Chemistry, Ural Branch RAS, Russia
In the context of the importance of finding new superconductors a significant interest on
recent discovery of superconductivity in ternary boride Ca3Rh8B6 with TC = 4 K [1] take place.
This boride includes in the homologous series AM3n-1B2n (A = Ca, Sr; M = Rh, Ir), n = 1, 2, 3,
which members have an orthorhombic structure. In addition to these phases, in ternary Ca-M-B
system, it is known about the existence of CaM3B2 (M = Rh, Ir) borides with hexagonal crystal
structure.
For the first time we have studied the main features of structural, electronic properties, phase
stability and Fermi surface topology of eight Ca containing ternary borides CaM2B2, CaM3B2,
Ca2M5B4, Ca3M8B6 (M = Rh, Ir) within FLAPW-GGA method realized in WIEN2k code [2].
We calculate the formation energies (Eform) of the borides based on the following reactions
such as Ca + 2M + α-B → CaM2B2. It is found that all values Eform <0 and are comparable with
each other, i.e. all borides are energetically stable with respect to mechanical mixture of Ca, M,
and α-B, see Table 1. In addition, Ir borides are more stable compared to Rh borides.
A characteristic feature of the electronic spectra for CaM2B2, CaM3B2, Ca2M5B4 and
Ca3M8B6 (M = Rh, Ir) is the presence of two main regions with different partial composition.
Fermi level crosses low-dispersion bands having predominantly M d nature and determining the
metallic properties of borides. Thus for CaM2B2, CaM3B2 near-Fermi region falls to the bottom
of the density of states, while for Ca2M5B4 and Ca3M8B6 the density of electronic states at the
Fermi level N(EF) significantly increases.
Table 1. The formation energies relatively to Ca, M and α-B (Eform, in eV/atom) for ternary
borides CaM2B2, CaM3B2, Ca2M5B4 и Ca3M8B6 (M=Rh, Ir).
boride CaRh2B2 CaRh3B2 Ca2Rh5B4 Ca3Rh8B6
Eform -0.817 -0.881 -0.856 -0.874
boride CaIr2B2 CaIr3B2 Ca2Ir5B4 Ca3Ir8B6
Eform -0.956 -0.917 -0.939 -0.942
[1] Takeya H, ElMassalami M, Terrasoz LA, Rapp RE, Capaz RB, Fujii H, Takano Y, Doerr M,
Granovsky SA. Sci. Techn. Adv. Mater. 2013; 14:035003.
[2] Blaha P, Schwarz K, Madsen GKH, Kvasnicka D, Liutz J, WIEN2k, An Augmented Plane
Wave Plus Local Orbitals Program for Calculating Crystal Properties, Vienna: Vienna
University of Technology, 2001.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–71–
OXYGEN ION DIFFUSIVITY IN SCANDIA-STABILIZED
ZIRCONIA: MOLECULAR DYNAMICS SIMULATIONS AND
AB INITIO CALCULATION
Maria Ivonina*, Pavel Snegurov, Vladimir Sizov
Saint Petersburg State University, Saint Petersburg, Russia
The cubic phase of zirconia (zirconium dioxide, ZrO2) stabilized by oxides of aliovalent
metals displays ionic conductivity caused by the presence of charge-balancing vacancies in the
anionic sub-lattice. Zirconia-based ionic conductors are widely used as electrolytes in solid
oxide fuel cells. In particular, doping of zirconia by scandium oxide (Sc2O3) results in scandia-
stabilized zirconia (ScSZ), which can achieve higher conductivity compared to other commonly
used materials under the same operating conditions [1]. In this work classical molecular
dynamics simulations and ab initio calculations are used to study the mechanisms of oxygen
diffusion in ScSZ.
The dependence of oxygen self-diffusion coefficients on temperature and scandia
concentration was studied by molecular dynamics simulations. The number of vacancies
increases with increasing dopant concentration, providing a wide variety of possible migration
pathways for oxygen ions. These migration pathways were analyzed by monitoring the
trajectories of oxygen ions obtained from molecular dynamics simulations. Investigation of the
dependence of diffusion coefficients on inverse temperature provides information on diffusion
activation energies, which can be used to quantify the energy barriers for diffusion of oxygen
ions.
Nudged elastic band (NEB) method was used as an alternative ab initio approach to evaluate
the energy barriers for various oxygen migration pathways in ScSZ. The results obtained from
ab initio calculations were compared to the data obtained from classical simulations and from
the literature [2–3].
T. H. Etsell, S. N. Flengas, Chem. Rev. 70, 339-376 (1970).
Z. Q. Yu, R. Devanathan, W. Jiang, P. Nachimuthu, V. Shutthanandan, L. Saraf, C. M.
Wang, S. V. N. T. Kuchibhatla, S. Thevuthasan, Solid State Ionics 181, 367–371 (2010).
A. Kushima, B. Yildiz, J. Mater. Chem. 20, 4809–4819 (2010).
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–72–
ATOMISTIC SIMULATION OF STACKING FAULTS IN CEMENTITE
Kar’kina L.E.a*, Kar’kin I.N. a,b, Kuznetsov A.R. a,b
aInstitute of Metal Physics UB RAS, Ekaterinburg, Russia,
bInstitute of Quantum Materials Science, Ekaterinburg, Russia
Precipitation of lamellar or globular cementite in ferrite defines high strength of pearlite class
steels. Experimental investigations have established that cementite can be deformed not only at
high temperatures, but also at room temperature. However, the experimental determination of
the slip systems in cementite encounters considerable experimental difficulties. Atomistic
simulation of stacking faults allows us to offer possible and the most energetically favorable
types of full and partial dislocations, which carry out the deformation of cementite.
Molecular dynamics method was used to study γ-surfaces for some planes of cementite
containing Burgers vectors [100] and [010]. Displacement vectors corresponding to stable
stacking faults have been determined. The energy of these stacking faults has been calculated by
the molecular dynamics and ab initio methods. The energy of unstable stacking faults, which
characterizes the tendency of a material to plastic relaxation, has been estimated. The reactions
of the splitting of perfect dislocations have been suggested; the possibility of the propagation of
stacking faults in the planes under consideration is discussed.
The results of crystallographic analysis of deformation transfer mechanisms across the
ferrite/cementite interface in fine lamellar pearlite are presented. Slip planes and Burgers vectors
of full and partial dislocations in cementite have been proposed on the basis of the results of
atomistic simulations of stacking faults for some planes of cementite. It has been found that
strain transfer through the Fe/Fe3C interface is possible only for half of the slip systems
1/2<111>{110}F of ferrite.
The research was carried out within the state assignment of FASO of Russia (themes
“Structure” No. 01201463331 and “Deformation” No. 01201463327). The results have been
obtained using the computational resources of MCC National Research Center “Kurchatov
Institute” (http://computing.kiae.ru) and Uran supercomputer of Institute of mathematics and
mechanics UB RAS.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–73–
THE PRECIPITATION KINETICS AND GPZ FORMATION IN AL-
BASED ALLOYS. MASTER EQUATION APPROACH WITH AB-INITIO
PARAMETERIZATION
Andrey Stroev1,2, O. Gorbatov2, Yuri Gornostyrev1,2
1Institute of Metal Physics Ural Branch RAS, Ekaterinburg, Russia
2Institute of quantum materials science, Ekaterinburg, Russia
AlCu-based alloys still nowadays are of great importance for light-weight and durable
constructions. The hardening of Al is crucially dependent on coherent meta-stable precipitates
formed during aging at room or moderately high temperature. In binary AlCu alloys these
precipitates are thin Cu platelets of a few nanometer thickness on the {100}-planes in fcc-Al;
they are called Guinier-Preston zones (GPZ).
The mixed-space cluster expansion (MSCE) model coupled with Monte Carlo
simulations was proposed in Ref. [1] to describe precipitation of Cu in fcc Al. Here we develop
consistent “flat-space” approach based on the recently developed master equation method [2] for
description of decomposition of binary alloys. In this approach the energy of alloy is presented
in the form of the cluster expansion taking into account as pair as well ternary and quaternary
interaction between solute atoms. Wherein, instead of the anisotropic elastic contribution to
interaction energies, we consider renormalization chemical interactions due to atomic relaxation.
A simple master equation based model with parameterization from ab-initio calculations for Al-
Cu and Al-Mg alloys is presented. Within framework of this approach, the numerical
simulations of alloy decomposition were done and the dependence of precipitates morphology
on temperature/concentration has been investigated. Based on obtained results, conditions of the
GPZ formation have been clarified.
The research was carried out within the state assignment of FASO of Russia (themes
“Structure” No. 01201463331) and under financial support from the Russian Science Foundation
(grant 14-12-00673).
[1] S. Muller, C. Wolverton, L.-W. Wang and A. Zunger, Europhys. Lett., 55, 33 (2001)
[2] K. Yu. Khromov, F. Soisson, A. Yu. Stroev, V. G. Vaks. Solid State Phenomena, 172-174,
1146-1155, (2011).
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–74–
DECOMPOSITION KINETICS IN Fe–Cu DILUTE ALLOYS.
MONTE CARLO SIMULATION
Ivan Shmakov1,2, Ilya Razumov1,2, Yuri Gornostyrev1,2
1 Institute of Metal Physics Ural Branch RAS, Russia
2Institute of quantum materials science, Russia
A significant effect of bcc Cu precipitates on the mechanical characteristics stimulated
numerous studies of the decomposition of supersaturated Fe–Cu based solid solutions using
advanced experimental techniques and theoretical approaches [1]. Here we propose a
generalization of the statistical (Monte Carlo) simulation technique for modeling of the
decomposition kinetics in the Fe–Cu system by taking into account the dependence of the
effective interactions both on the magnetic state of the alloy and on the local concentration of the
dissolved chemical element. Similar approach was early implemented in [2] for the Fe–Cr alloy.
We demonstrate that the concentration dependence of the effective interactions results in
significantly shift the line corresponding to the onset of the transformation in the time–
temperature– transformation (TTT) diagram toward longer times providing better agreement
with the experiment. Thus, the presented results demonstrate that the inclusion of the
concentration dependence of the effective interactions is critically important for the adequate
description of the decomposition kinetics in the alloy [4].
Fig. 1. Calculated TTT diagram of decomposition the dilute Fe–1.2 at %Cu alloy. The curves
correspond to achieving the specified degree of decomposition of 0.10 (curves 1 and 2) and 0.20
(curves 1' and 2') at a fixed temperature. The concentration dependence of the effective
interactions is neglected (curves 1 and 1') and taken into account in curves 2 and 2'. Curve 3
shows the experimental data [3] for a transformation fraction of 10%.
[1] O. I. Gorbatov, I. K. Razumov, Yu. N. Gornostyrev, V. I. Razumovskiy, P. A.
Korzhavyi, and A. V. Ruban, Phys. Rev. B 88, 174113 (2013).
[2] A. V. Ruban, P. A. Korzhavyi, and B. Johansson, Phys. Rev. B 77, 94436 (2008)
[3] M. Perez, F. Perrard, V. Massardier, X. Kleber, A. Deschamps, H. De Monestrol, P.
Pareige, and G. Covarel, Philos. Mag. 85, 2197 (2005)
[4] I.G. Shmakov, I.K. Razumov, O.I. Gorbatov, Yu.N. Gornostyrev, P.A. Korzhavyi,
ZhETP Letters, 103, No. 2, 119 (2016)
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–75–
MOLECULAR DYNAMICS SIMULATION OF BULK XENON
DIFFUSION IN UO2: A COMPARISON OF AB INITIO INTERACTION
POTENTIALS
Kirill Nekrasov*, Natalia Kichigina
Department of Technical Physics, Ural Federal University,
620002 Yekaterinburg, Russia
Accumulation of radiogenic xenon in nuclear fuel causes swelling of the material and can
degrade its exploitation characteristics. Transport of xenon atoms in the oxide fuels such as UO2
can be studied using molecular dynamics simulation. A number of sets of ab initio interaction
potentials for the Xe – UO2 system have been suggested, some of them calculated using the
relativistic Hartree-Fock method [1] and other fitted to density functional theory calculations [2–
4]. In this work, the potentials [1–2, 4] are used for molecular dynamics simulation of bulk
migration of single Xe atoms in stoichiometric UO2 nanocrystals with free surface. The diffusion
coefficients and activation energies and mechanisms obtained for the different potential sets are
compared with each other and the existing experimental data. The calculated values of the
diffusion coefficients and energies are in quantitative agreement with the experiment.
[1] Jackson R A and Catlow C R A 1985 J. Nucl. Mater. 127 161.
[2] Geng H Y, Chen Y, Kaneta Y and Kinoshita M 2008 J. Alloys Compounds 457 465.
[3] Chartier A, Van Brutzel L and Freyss M 2010 Phys. Rev. B 81 174111.
[4] Thompson A E, Meredig B and Wolverton C 2014 J. Phys.: Condens. Matter 26 105501.
*Email: [email protected]

AB INITIO BASED MODELING OF ADVANCED MATERIALS 2016
–76–
SIZE MISFIT VERSUS ELECTRONIC EFFECTS IN DIFFUSION OF
SUBSTITUTIONAL IMPURITIES IN NI MATRIX
Ilya Lomaev1,2*, Dmitry Novikov3, Sergey Okatov1,
Yuri Gornostyrev1,2, Sergey Burlatsky3
1 CJSC Institute of Quantum Materials Science, Yekaterinburg, Russia
2 Institute of Metal Physics UB RAS, Yekaterinburg, Russia 3 United Technologies Research Center (UTRC), USA
Atomic diffusion is one of the key factors controlling phase transformations and
microstructure evolution in materials, especially at high temperatures. Despite the obvious
fundamental and technological importance of diffusion in solids, we still know little about the
microscopic mechanisms and factors that affect it. In the present study we implement a
transition state theory of vacancy mediated diffusion with parameters calculated from first-
principle density-functional theory (DFT) to determine the diffusion coefficients of SP and
transition metal (TM) substitutional impurities in fcc Ni.
According to the traditional point of view, a larger atom has larger migration barrier and
consequently lower diffusion mobility. We demonstrate that this is not the case for diffusion of
TM elements in Ni matrix. The larger atoms indeed produce larger lattice strain. However, this
results firstly in the higher solute–vacancy binding energy. Secondly, the equilibrium position of
impurity becomes shifted towards adjacent vacancy. The larger atoms are shifted strongly, with
their migration barriers being significantly decreased. These two reasons lead to a controversial
at first site consequence — the larger atoms diffuse faster. Nevertheless, this phenomenon still
can be described using size misfit arguments.
Quite different situation is observed in the case of SP impurities. We demonstrate that
diffusivities of SP-impurities in TM-matrix are mainly affected by features of chemical bonding
rather than by atomic size misfit and are well correlated with charge on the impurity atom. As an
example, the sulfur atom has the lowest charge among other SP-impurities and as a result its
diffusion coefficient was found to be at list two orders of magnitude higher than that for others
in good agreement with experimental data. Moreover, sulfur was shown to have very low
migration barrier due to formation of covalent-like bonds between S3p and Ni3d orbitals in
saddle point and destabilization (“softening”) of the surrounding Ni-matrix.
This work was performed according to the State Task of the FANO of the Russian Federation
(theme “Deformation,” no. 01201463327)
*Email: [email protected]

AUTHOR INDEX
Andreev Sergey .................................................................................... 8,51
Anisimov Vladimir ......................................................................... 3,16,29
Badrtdinov Danis ............................................................................ 6,9,57,64
Bannikov Vyacheslav ........................................................................... 8,54
Barbin Nikolay...................................................................................... 9,63
Boukhvalov Danil ........................................................................... 6,9,36,69
Chernyshev Vladimir ................................................................ 8,9,58,66,67
Eriksson Olle ........................................................................................ 3,17
Gorbatov Oleg .................................................................................. 6,41,73
Gornostyrev Yuri ...................................................... 10,36,41,45,73,74,76
Gerasimov Arsenii ................................................................................ 7,49
Greshnyakov Vladimir ......................................................................... 7,47
Hickel Tilmann ............................................................................... 5,6,31,33
Ivonina Maria...................................................................................... 10,71
Kashin Iliya ........................................................................................... 8,50
Kardashin Andrey ...................................................................................... 10
Karkina Lydia ....................................................................................... 10,72
Katanin Andrey ........................................................................ 3,8,13,59,60
Katsnelson Mikhail ........................................................................... 9,36,61
Khmelevskyi Sergei .............................................................................. 5,30
Komleva Evgenia.................................................................................. 8,56
Korotin Mikhail ...................................................................................... 5,26
Korzhavyi Pavel...................................................................................... 7,43
Kuznetsov Andrey ................................................................................ 10,72
Leonidov Ivan ......................................................................................... 9,67
Leonov Ivan ...................................................................................... 3,14,29
Lichtenstein Alexander ................................................................... 3,8,12,55
Lomaev Ilya .................................................................................... 10,41,76
Mazurenko Vladimir ............................... 4,7,9,10,25,49-52,55-57,61,68,69
Medvedeva Daria .................................................................................... 8,55
Medvedeva Nadezhda ............................................................................. 6,40
Mirzoev Alexander ................................................................................. 6,39
Morris James ........................................................................................... 7,42
Nazipov Dmitry ...................................................................................... 9,65
Nekrasov Igor ................................................................................. 4,8,19,53
Nekrasov Kirill ..................................................................................... 10,75
Neugebauer Jörg ............................................................................ 5,6,31,33
Oguchi Tamio ......................................................................................... 4,23
Pavlov Nikita .......................................................................................... 8,53
Pchelkina Zlata ....................................................................................... 9,68
Petrik Mikhail ................................................................................. 7,9,45,64
Piterskikh Ilya ......................................................................................... 9,69
Poteryaev Alexander ............................................................................. 5,32
Pourovskii Leonid ................................................................................. 3,18

–78–
Pravednicov Andrey .................................................................................. 10
Prishchenko Danil ............................................................................. 9,10,61
Protsenko Vladimir ............................................................................... 8,60
Razumov Ilya ............................................................................................. 74
Razumovskiy Vsevolod ........................................................................ 6,38
Rohringer Georg ................................................................................... 4,21
Rubtsov Alexey .................................................................................... 4,22
Rudenko Alexander ........................................................................ 6,9,27,61
Savrasov Sergey.................................................................................... 5,35
Sekania Mikheil .................................................................................... 7,44
Shmakov Ivan ..................................................................................... 10,74
Shundalov Maksim ............................................................................... 7,48
Simak Sergey ........................................................................................ 6,37
Skornyakov Sergey ......................................................................... 5,7,29,49
Slobodchikov Anatoliy ......................................................................... 8,53
Solovyev Igor......................................................................... 4,7,8,24,50,51
Sotnikov Oleg ....................................................................................... 8,52
Sozykin Sergey ..................................................................................... 9,62
Stepanenko Andrey ............................................................................... 8,59
Streltsov Sergey .................................................................................... 3,15
Stroev Andrey ..................................................................................... 10,73
Suetin Dmitry........................................................................................ 9,70
Toschi Alessandro ............................................................................ 4,20,21
Tsirlin Alexander ..................................................................... 5,8,10,28,56
Verkhovykh Anastasia .......................................................................... 6,39
Vesnina Daria ....................................................................................... 8,59
Zainullina Veronika ...................................................................................
Zakir’yanov Dmitry .............................................................................. 8,58

–79–

–80–

–81–

–82–

–83–

–84–
Научное издание
ПЕРВОПРИНЦИПИАЛЬНОЕ МОДЕЛИРОВАНИЕ ПЕРСПЕКТИВНЫХ
МАТЕРИАЛОВ
Рекомендовано к изданию Ученым Советом ИФМ УрО РАН
и Президиумом УрО РАН
Редактор
Технический редактор
Компьютерная верстка
Дизайн
Подписано в печать
Тираж 80 экз
Издательство
Отпечатано в типографии