a theoretical study of the catalytic mechanism of oxalyl ...210.75.249.4/bitstream/363003/4272/1/a...
TRANSCRIPT

RSC Advances
PAPER
A theoretical stu
aSchool of Chemistry and Chemical En
Shandong 250100, ChinabNorthwest Institute of Plateau Biology, Chin
810001, China. E-mail: [email protected]
531 883 655 76cSchool of Agriculture, Ludong University, Y
† Electronic supplementary informationproton transfer process from 40-NH2 to tas the starting structures, respectively. Se
Cite this: RSC Adv., 2014, 4, 35777
Received 21st April 2014Accepted 4th August 2014
DOI: 10.1039/c4ra03611e
www.rsc.org/advances
This journal is © The Royal Society of C
dy of the catalytic mechanism ofoxalyl-CoA decarboxylase, an enzyme for treatingurolithiasis†
Xiang Sheng,a Yongjun Liu*ab and Rui Zhangc
Oxalate is harmful to many organisms by forming insoluble precipitates with some metal cations. In
humans, calcium oxalate is a major constituent of kidney stones leading to urolithiasis. Oxalobacter
formigenes is a bacterium in most vertebrates and can regulate the homeostasis of oxalate. Replacement
therapies of O. formigenes or related-enzymes are new strategies for treating oxalate-related diseases.
Oxalyl-CoA decarboxylase (OXC) is an enzyme involved in the oxalate degradation in O. formigenes. In
this paper, the catalytic mechanism of OXC was investigated by using the density functional theory (DFT)
method. The most likely reaction pathway, detail of elementary steps, and roles of key residues were
determined. Our calculation results indicate that the decarboxylation process can proceed rapidly, which
agrees well with the experimental observation. In the protonation of the HDC-ThDP intermediate, the 4-
NH2 of ThDP is suggested to be the proton donor, which abstracts a proton from the nearby residue
E121. The rate-limiting step is calculated to be the proton transfer from 40-NH2 to the HDC-ThDP
intermediate with an energy barrier of 21.8 kcal mol�1. However, if this pathway is blocked by mutating
residue E121, the reaction may follow another mechanism, in which Y483 acts as the proton donor and
uses a water molecule as a mediator. These findings can explain the experimental observation that
replacement of residues Y483 or E121 significantly reduces but does not completely abolish the activity
of the enzyme. Our results may provide useful information for exploring the enzymatic mechanism and
developing biocatalytic applications for treating the oxalate-related diseases.
1. Introduction
Oxalate is a compound that is naturally present in animals,plants and fungi. It can form insoluble precipitates with somemetal cations, causing oxalate to be ecologically harmful tomany organisms.1 In humans, calcium oxalate is a majorconstituent of kidney stones2 leading to urolithiasis. Theincrease of oxalate level has also been demonstrated to beassociated with renal failure,3 cardiomyopathy4 and cardiacconductance disorders.5 However, humans and other mammalsare unable to biodegrade oxalate, and this compound can onlybe excreted through urine or absorbed by intestine.6 As oxalateis mainly acquired from diets, one strategy to prevent oxalate-related diseases is to cut down the daily consumption offoods rich in oxalate,7 but its long-term effectiveness is still
gineering, Shandong University, Jinan,
ese Academy of Sciences, Xining, Qinghai
du.cn; Fax: +86 531 885 644 64; Tel: +86
antai, Shandong 264025, China
(ESI) available: The energy prole ofhe hydroxyl anion using IM2 and IM3e DOI: 10.1039/c4ra03611e
hemistry 2014
uncertain and such a diet would probably cause the decienciesof some essential nutrients.8
Recently, an obligate anaerobe and gut-dwelling bacterium,Oxalobacter formigenes, has been proved to be directly relevantto the recurrence of kidney stone episodes.9 O. formigenesmainly exists in the gastrointestinal tracts of most vertebratesincluding humans, and uses oxalate as the sole energy sourcefor their survival,10 which means O. formigenes can be used toregulate the homeostasis of oxalate, providing a new strategy fortreating oxalate-related diseases. The lack of O. formigenes couldincrease the excretion of urinary oxalate and promote theformation of calculus.11 Strong evidences have supported thatpatients of urolithiasis with O. formigenes in their stool canlower the oxalate excretion in their urine.9b,12 Furthermore,traditional treatments are scarce and only achieve satisfactoryresults in a minority of patients.13 Thus, replacement therapiesof O. formigenes or related-enzymes have attracted much atten-tion,14 and colonization of O. formigenes has been proved to beeffective in decreasing urinary oxalate levels15 and reducing therisk of stone recurrence.16 However, clinical studies showed thatthis colonization is not a permanent solution,14a which inspiresinterest to develop the enzyme-based therapies.
Two enzymes, formyl-CoA transferase (FRC)17 and oxalyl-CoAdecarboxylase (OXC),18 are involved in the oxalate anaerobic
RSC Adv., 2014, 4, 35777–35788 | 35777

Scheme 1 The catalytic reaction of oxalyl-CoA decarboxylase (OXC).
RSC Advances Paper
degradation in O. formigenes. As shown in Scheme 1, FRC rstlycatalyzes the transfer of a CoA from formyl-CoA to oxalate toactivate the subsequent thiamin-dependent decarboxylation ofoxalyl moiety. The formed oxalyl-CoA is then decarboxylated toproduce CO2 and regenerate formyl-CoA by the catalysis of OXC,a thiamin diphosphate (ThDP)-dependent nonoxidative decar-boxylase. As proposed in the ref. 18, the catalytic cycle can bedivided into two half-reactions with four steps, as shown inFig. 1: (1) aer the activation of ThDP, the covalent substrate–ThDP adduct (a-oxalyl-CoA-ThDP intermediate) is rstly formedby the attack of C2 carbanion of ThDP ylide on the carbonylgroup of substrate oxalyl-CoA; (2) the scissile C2a–C2b bond of a-oxalyl-CoA-ThDP is then broken to form a a-hydroxyl-CoA-ThDPcarbanion/enamine intermediate (HDC-THDP), and CO2 isliberated; (3) the C2a atom of HDC-THDP accepts a proton toyield a-formyl-CoA-ThDP adduct, and (4) the C2–C2a bond iscleavaged to regenerate formyl-CoA.
Recently, Lindqvist et al. obtained several crystal structuresof OXC from O. formigenes,18a,b in which the diphosphate and 30-phosphate of CoAmoiety of substrate are positioned by residuesR266, R408 and R555, and the oxalyl part interacts directly or viaa bridging water molecule with residues I34, Y120 and Y483.The cofactor ThDPmaintains its V-like conguration, which hasbeen proved to be prerequisite for catalysis.19 A conservedglutamate residue (E56 in OXC from O. formigenes) forms ahydrogen bond with the 10-N of ThDP, and is considered to playan important role in the activation process of ThDP.20 In addi-tion, the 40-NH2 of ThDP forms two hydrogen bonds with resi-dues Y120 and E121. Besides, some mutant experiments havebeen performed for understanding the catalysis of this enzy-me.18b For example, replacements of residues E56, Y120, E121,Y483 or S553 greatly reduced the activity of enzyme, but thesemutants were not completely inactive, except E56A. Based onthese structural and kinetic data, Lindqvist et al. proposed thatno pocket residues directly participate in the proton transferaer ThDP activation, but a water molecule protonates theHDC-ThDP intermediate.
At present, a rough picture of the catalytic mechanism of OXChas been obtained. But open questions still remain unresolved.For example, a water molecule was suggested to protonate theHDC-ThDP intermediate, and this water molecule exists in theform of hydroxide anion in the post-decarboxylation interme-diate complex.18a,b It has been known that, in some enzymes Zn2+
cation serves as Lewis acid to assist the water molecule to donate
35778 | RSC Adv., 2014, 4, 35777–35788
its proton.21 But in OXC, no Zn2+ cation or other strong Lewisacid exists in the catalytic site. Thus, it is interesting to knowwhich residue plays this role. Besides, in some other ThDP-dependent enzyme, the 40-NH2 of ThDP can function as ageneral acid/base to protonate the carbonyl group of substrate.22
Is it possible that the HDC-ThDP intermediate is protonated bythe 40-NH2? In addition, the specic roles of key residues in theactive site, and the energetic information about the bondformation and cleavage are still not understood. An explicitdescription of the enzymatic mechanism is of particularlysignicance for exploring the enzymatic characterization anddeveloping biocatalytic applications, and some valuable infor-mation cannot be acquired by experiments alone. Therefore,theoretical studies on the reaction mechanism at the atomisticlevel are still necessary. However, in contrast to the extensivestudies on some other ThDP-dependent enzymes, such aspyruvate decarboxylase (PDC),23 acetohydroxyacid synthase(AHAS)24 and oxidoreductase (PFOR),25 so far there is no theo-retical study on the catalytic mechanism of OXC.
Here, we present a density functional theory (DFT) study onthe catalytic mechanism of OXC with cluster approach. In thepast decades, the cluster approach has been widely applied inqualitatively elucidating the enzymatic reaction mechanisms.26
It should be noted that the combined quantum mechanics andmolecular mechanics (QM/MM) method, which has beenincreasingly applied for investigating the fundamental prob-lems of enzymology,27 should be the preferred strategy for thisstudy. However, studies on different types of enzymes by usingcluster approach have also shown that the calculation resultswith relative large cluster models were reliable and useful forqualitatively elucidating the enzymatic reaction mechanisms.28
In addition, the cluster approach is easy to operate, therefore,the cluster approach was selected in the present study. Based onour calculations, the reaction pathways, the detailed energeticproles of the entire reaction and the structures of involvedspecies are presented, and the nature of proton donors and theroles of key pocket residues were determined as well.
2. Computational methods2.1 Cluster models
The cluster models used in this work are constructed on thebasis of recently obtained crystal structure of OXC from O. for-migenes (PDB code: 2JI7),18b which is a covalent HDC-ThDPintermediate aer the decarboxylation of the a-oxalyl-CoA-ThDP intermediate. From the active site structure shown inFig. 2, we can see that in this post-decarboxylation intermediate,a water molecule (W1) has taken the place of the previouslyformed CO2. Thus, it can be used to construct the computa-tional model for investigating the second half-reaction. Theselected atoms in our model are shown in ball and stick inFig. 2, which contain the decarboxylated substrate–ThDPadduct HDC-ThDP, two water molecules (W1 andW2) and somekey residues including E56, E121 and G426, which formhydrogen bonds with the ThDP moiety, and I34, Y120 and Y483that interact with the CoA part directly or via water molecules.
This journal is © The Royal Society of Chemistry 2014

Fig. 1 Proposed catalytic mechanism of OXC.
Fig. 2 Crystal structure of the active site of OXC (PDB code: 2JI7) in apost-decarboxylation intermediate form.
Paper RSC Advances
Due to the crystal structure of OXC in complex with thesubstrate a-oxalyl-CoA and cofactor ThDP is still not available,according to the principle of microreversibility, we reversiblystudied the rst half-reaction by using an intermediate that thedecarboxylation has been completed. To construct the startingstructure for studying the rst half-reaction, the water moleculeW1 in the crystal structure (PDB code: 2JI7) was removed andthe CO2 molecule was added manually. Thus, in fact, the initialstructure for the rst half-reaction is IM2, and that for thesecond half-reaction is IM3, as shown in Fig. 3.
The protonation states of all ionizable residues involved inthe cluster models were determined according to the experi-mental condition, and their pKa values were predicted by thePROPKA 3.1 program.29 Aer adding hydrogen atoms, both oftwo cluster models contain 133 atoms. In our calculations, theHDC-THDP and all residues were truncated so that onlyimportant peptide backbones or side chains were included.During the optimizations, the truncated atoms were kept xed
This journal is © The Royal Society of Chemistry 2014
to their crystallographic positions to prevent unrealistic move-ments of the groups, and the xed atoms were marked withasterisks in the corresponding gures. It can be seen that only afew atoms were xed in the models during the optimizations,including the terminal carbon atoms of truncated CoA part andresidues I34, E56, Y120, E121, G426, and Y483 as well as theterminal carbon and oxygen atoms of truncated ThDP moiety.
Since there are extensive experimental and computationalinvestigations on the activation mechanism of ThDP else-where,23a,30 the activation process was not considered in ourcalculations. Based on the active site of used crystal structure (PDBcode: 2JI7), we assume that residue Y483 may act as the acid todonate its proton of phenolic hydroxyl group to the watermoleculeW1, which has already donated its proton to C2a atom of HDC-ThDP intermediate. In addition, the 40-NH2 may also act as theproton donor. Thus, for the second half-reaction, according to thedifferent proton donors, two reaction pathways (Path-1 and Path-2) were considered, as shown in Fig. 3, in which the water mole-cule W1 and the 40-NH2 respectively serve as the proton donor. InPath-2, aer the proton transfer from 40-NH2 of ThDP to C2a atom,the reaction will undergo two elementary reactions, including theproton transfer from E121 to 40-NH, and the cleavage of C2–C2a
bond. Since the proton transfer process may occur prior or later tothe cleavage of C2–C2a bond, therefore, two possible reactionpathways (Path-2A and Path-2B) were considered for Path-2.
2.2 Computational details
All quantum chemical calculations presented here were carriedout by employing the Gaussian 09 program package.31 Allgeometrical optimizations were conducted with the hybridmeta-GGA density functional M06-2X32 and the 6-31G(d,p) basis set,and all single-point calculations were performed with the largerbasis set 6-311++G(2d,2p). To consider the effect of surroundingprotein environment on the energetics that was not included inthe computational models, we used the polarizable-continuummodel (PCM)33 to calculate the single-point energies at thesame level for each species on the above optimized geometries.In this solvation model, the solvent is represented by a constantdielectric medium surrounding a cavity containing the solute.The empirical dielectric constant of the enzyme environment ischosen to be 4, which has been used in many studies.34
Harmonic frequency calculations were performed with the 6-
RSC Adv., 2014, 4, 35777–35788 | 35779

Fig. 3 The proposed reaction pathways catalyzed by OXC in our calculations. For the second half-reaction, according to the different protondonors, two reaction pathways (Path-1 and Path-2) are considered. In path-2, two cases were studied, in which the proton transfer from E121 to40-NH can occur prior (Path-2A) or later (Path-2B) than the cleavage of C2–C2a bond.
RSC Advances Paper
31G(d,p) basis set to obtain zero-point vibrational energies (ZPE)and to conrm the nature of stationary points. Forcing thetruncation atoms to their crystallographic positions during thegeometrical optimizations gives rise to a few small negativeeigenvalues, typically in the order of 10 cm�1. These frequenciesare very small and do not contribute signicantly to the zero-point energies and thus can be tolerated. Intrinsic reactioncoordinate (IRC) calculations were preformed to conrmconnection of transition states to two relevant minima.35
3. Results and discussion3.1 Reaction pathways
For ThDP, the characteristic torsion angles FT dened by atomsC50–C3, 50-N3–C2 and FP dened by C40–C50–C3, 50-N3 have
35780 | RSC Adv., 2014, 4, 35777–35788
been suggested to be important parameters for describing therelative orientation of the two rings.36 Tables 1 and 2 list thevalues of FT and FP of all species, which indicate that the so-called “V” conformation of ThDP is well kept during the cata-lytic reaction. To clearly elucidate the catalytic cycle, the rstand second half-reactions are respectively discussed in thefollowing sections. All reported energies are obtained at thelevel of M06-2X/6-311++G(2d,2p)//M06-2X/6-31G(d,p) in thesolvent phase (PCM, 3 ¼ 4).
3.1.1 The rst half-reaction. Although the calculationswere started from the intermediate IM2 and the correspondingreactant and transition state structures were derived from IM2,to conveniently elucidate the reaction mechanism, we stilldiscuss the rst half-reaction by the forward order, i.e., theligation of C2 atom of ThDP and C2a atom of substrate oxalyl-
This journal is © The Royal Society of Chemistry 2014

Table 1 The characteristic torsion angles FT and FP (�) for theorientation of the two ThDP rings, and the lengths (A) of some keyhydrogen bonds in the species of the first half-reaction
R TS1 IM1 TS2 IM2
FP �79 �74 �65 �68 �69FT 74 100 111 109 105dW2-I34 2.13 2.26 2.15 2.18 2.12dW2-Y120 2.03 1.92 1.96 1.89 1.83dW2-E121 2.00 2.09 1.996 2.02 2.03dY120-O 1.86 1.73 1.59 1.56 1.48dE56-10 -N 1.71 1.73 1.73 1.70 1.67dG426-40 -NH2 1.96 1.93 1.93 1.96 2.00
Table 2 The characteristic torsion angles FT and FP (�) for theorientation of the two ThDP rings, and the lengths (A) of some keyhydrogen bonds in the species of Path-1 and Path-2 of the secondhalf-reaction
Path-1 IM3 TS3-1 IM4-1 TS4-1 P-1
FP �69 �64 �59 �77 �83FT 104 109 115 92 71dW2-I34 2.06 0.11 2.11 2.15 2.17dW2-Y120 1.78 1.81 1.87 1.86 1.85dW2-E121 2.02 2.03 2.02 2.20 2.28dY120-O 1.43 1.41 1.47 1.62 1.69dE56-10 -N 1.65 1.68 1.70 1.67 1.72dG426-40 -NH2 1.97 1.97 2.01 1.95 1.97
Path-2A IM3 TS3-2 IM4-2 TS4-2A IM5-2A TS5-2A P-2A
FP �69 �70 �79 �86 �90 �86 �84FT 104 100 107 106 102 102 105dW2-I34 2.06 2.12 2.11 2.09 2.26 2.59 2.32dW2-Y120 1.78 1.77 1.78 2.10 2.67 2.64 2.36dW2-E121 2.02 2.13 2.20 1.92 1.82 1.57 1.65dY120-O 1.43 1.43 1.50 1.80 1.91 2.00 1.90dE56-10 -N 1.65 1.61 1.60 1.65 1.67 1.65 1.62dG426-40 -NH2 1.97 2.12 2.12 2.26 2.40 2.48 2.14
Path-2B IM3 TS3-2 IM4-2 TS4-2B IM5-2B TS5-2B P-2B
FP (�) �69 �70 �79 �85 �83 �85 �69FT (�) 104 100 107 104 118 112 96dW2-I34 2.06 2.12 2.11 2.61 2.02 2.44 2.54dW2-Y120 1.78 1.77 1.78 2.39 2.01 2.49 3.08dW2-E121 2.02 2.13 2.20 2.02 2.06 1.81 142dY120-O 1.43 1.43 1.50 1.96 1.71 1.90 2.12dE56-10 -N 1.65 1.61 1.60 1.57 1.56 1.65 1.62dG426-40 -NH2 1.97 2.12 2.12 2.30 2.33 2.27 1.91
Fig. 4 Optimized geometry of the reactant R. The hydrogen bonds areshown with bond length in angstrom. The fixed terminal atoms in themodel are labeled by asterisks.
Fig. 5 Optimized geometries at the M06-2X/6-31G(d,p) level forvarious species involved in first half-reaction. The key bond distancesare shown in angstrom.
Paper RSC Advances
CoA occurs rstly, then the cleavage of C2a–C2b bond follows, asshown in Fig. 3.
The optimized structure of reactant R is shown in Fig. 4, inwhich many hydrogen bonds are formed among the watermolecule W2, cofactor ThDP, substrate oxalyl-CoA and pocketresidues. Specically, the carboxyl and carbonyl groups ofoxalyl-CoA form two hydrogen bonds with residues Y483 andY120; W2 forms three hydrogen bonds with the amino group ofI34 backbone, carboxyl group of E121 side chain and phenolic
This journal is © The Royal Society of Chemistry 2014
hydroxyl group of Y120 side chain; the 40-NH2 of ThDP formstwo hydrogen bonds with residues E121 and G426, and 10-Natom of ThDP forms one hydrogen bond with residue E56. Allthese hydrogen bond lengths are shorter than 2.2 A, which mayplay important role in stabilizing the intermediates and tran-sition states in the subsequent reaction.
The optimized structures of reactant (R), intermediates (IM1and IM2) and transition states (TS1 and TS2) are shown inFig. 5. The reactive C2 atom of ThDP thiazolium ring positions adistance (r1) of 2.96 A from the C2a of oxalyl-CoA, implying thisstructure is favorable for the covalent addition of substrate withThDP. From R to TS1, r1 is shortened from 2.96 A to 2.12 A.Downhill from TS1 to IM1, this distance is further decreased to1.54 A, indicating the a-oxalyl-CoA-ThDP intermediate has beenformed, which can be characterized by the sp3 hybridized C2a
carbon atom and bond length of C2a–O2a (1.33 A). In IM1, thehydrogen bonds (shown in Fig. 5) between Y483 and thecarboxyl group of oxalyl-CoA part are well kept, which meansthat Y483 may play signicant role in the following decarbox-ylation. Pervious experimental studies have suggested that inthe adduct of ThDP and oxalyl-CoA (IM1), the carbonyl oxygenatom had abstracted a proton from the 40-NH2 of ThDP which
RSC Adv., 2014, 4, 35777–35788 | 35781
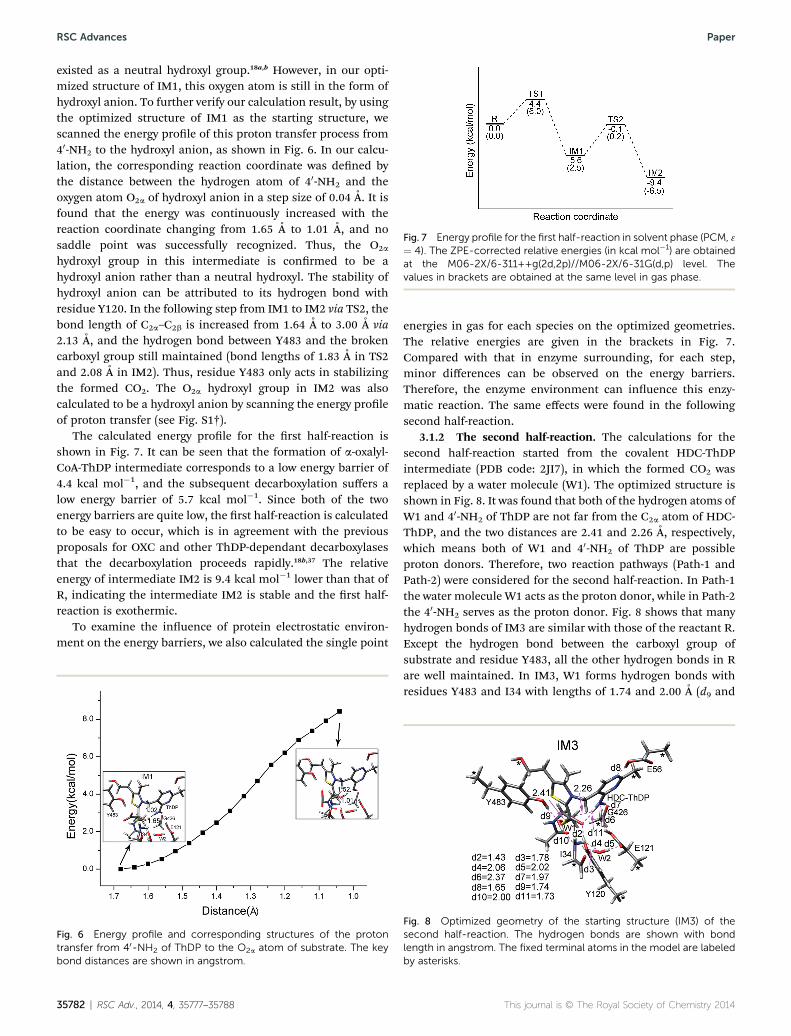
Fig. 7 Energy profile for the first half-reaction in solvent phase (PCM, 3¼ 4). The ZPE-corrected relative energies (in kcal mol�1) are obtainedat the M06-2X/6-311++g(2d,2p)//M06-2X/6-31G(d,p) level. Thevalues in brackets are obtained at the same level in gas phase.
RSC Advances Paper
existed as a neutral hydroxyl group.18a,b However, in our opti-mized structure of IM1, this oxygen atom is still in the form ofhydroxyl anion. To further verify our calculation result, by usingthe optimized structure of IM1 as the starting structure, wescanned the energy prole of this proton transfer process from40-NH2 to the hydroxyl anion, as shown in Fig. 6. In our calcu-lation, the corresponding reaction coordinate was dened bythe distance between the hydrogen atom of 40-NH2 and theoxygen atom O2a of hydroxyl anion in a step size of 0.04 A. It isfound that the energy was continuously increased with thereaction coordinate changing from 1.65 A to 1.01 A, and nosaddle point was successfully recognized. Thus, the O2a
hydroxyl group in this intermediate is conrmed to be ahydroxyl anion rather than a neutral hydroxyl. The stability ofhydroxyl anion can be attributed to its hydrogen bond withresidue Y120. In the following step from IM1 to IM2 via TS2, thebond length of C2a–C2b is increased from 1.64 A to 3.00 A via2.13 A, and the hydrogen bond between Y483 and the brokencarboxyl group still maintained (bond lengths of 1.83 A in TS2and 2.08 A in IM2). Thus, residue Y483 only acts in stabilizingthe formed CO2. The O2a hydroxyl group in IM2 was alsocalculated to be a hydroxyl anion by scanning the energy proleof proton transfer (see Fig. S1†).
The calculated energy prole for the rst half-reaction isshown in Fig. 7. It can be seen that the formation of a-oxalyl-CoA-ThDP intermediate corresponds to a low energy barrier of4.4 kcal mol�1, and the subsequent decarboxylation suffers alow energy barrier of 5.7 kcal mol�1. Since both of the twoenergy barriers are quite low, the rst half-reaction is calculatedto be easy to occur, which is in agreement with the previousproposals for OXC and other ThDP-dependant decarboxylasesthat the decarboxylation proceeds rapidly.18b,37 The relativeenergy of intermediate IM2 is 9.4 kcal mol�1 lower than that ofR, indicating the intermediate IM2 is stable and the rst half-reaction is exothermic.
To examine the inuence of protein electrostatic environ-ment on the energy barriers, we also calculated the single point
Fig. 6 Energy profile and corresponding structures of the protontransfer from 40-NH2 of ThDP to the O2a atom of substrate. The keybond distances are shown in angstrom.
35782 | RSC Adv., 2014, 4, 35777–35788
energies in gas for each species on the optimized geometries.The relative energies are given in the brackets in Fig. 7.Compared with that in enzyme surrounding, for each step,minor differences can be observed on the energy barriers.Therefore, the enzyme environment can inuence this enzy-matic reaction. The same effects were found in the followingsecond half-reaction.
3.1.2 The second half-reaction. The calculations for thesecond half-reaction started from the covalent HDC-ThDPintermediate (PDB code: 2JI7), in which the formed CO2 wasreplaced by a water molecule (W1). The optimized structure isshown in Fig. 8. It was found that both of the hydrogen atoms ofW1 and 40-NH2 of ThDP are not far from the C2a atom of HDC-ThDP, and the two distances are 2.41 and 2.26 A, respectively,which means both of W1 and 40-NH2 of ThDP are possibleproton donors. Therefore, two reaction pathways (Path-1 andPath-2) were considered for the second half-reaction. In Path-1the water molecule W1 acts as the proton donor, while in Path-2the 40-NH2 serves as the proton donor. Fig. 8 shows that manyhydrogen bonds of IM3 are similar with those of the reactant R.Except the hydrogen bond between the carboxyl group ofsubstrate and residue Y483, all the other hydrogen bonds in Rare well maintained. In IM3, W1 forms hydrogen bonds withresidues Y483 and I34 with lengths of 1.74 and 2.00 A (d9 and
Fig. 8 Optimized geometry of the starting structure (IM3) of thesecond half-reaction. The hydrogen bonds are shown with bondlength in angstrom. The fixed terminal atoms in the model are labeledby asterisks.
This journal is © The Royal Society of Chemistry 2014

Fig. 10 Energy profile for Path-1 of second half-reaction in solventphase (PCM, 3¼ 4). The ZPE-corrected relative energies (in kcal mol�1)are obtained at the M06-2X/6-311++G(2d,2p)//M06-2X/6-31G(d,p)level. The values in brackets are obtained at the same level in gasphase.
Paper RSC Advances
d10), respectively, and the hydrogen bond between 40-NH2 ofThDP and hydroxyl oxygen atom O2a is kept with a distance of1.73 A (d11). In addition, the existing form of C2a hydroxyl groupwas also explored by scanning the energy prole (see Fig. S2†). Itis demonstrated that the C2a hydroxyl group also exits in theform of hydroxyl anion.
The optimized structures of intermediate (IM4-1), transitionstates (TS3-1 and TS4-1) and product (P-1) involved in Path-1 areshown in Fig. 9. For a clear view of the structural changes, IM3was also included in the gure. In IM3, as mentioned above, theO–H bond of W1 is located in a favorable orientation for itshydrogen transfer to the C2a atom of HDC-ThDP, and thedistance (r6) between the hydrogen atom of W1 and the C2a
atom of HDC-ThDP is 2.41 A, which agrees with that in thecrystal structure with r6 of 2.70 A.18b In transition state TS3-1, r6is shortened to 1.35 A and the length (r5) of O–H bond of W1 iselongated to 1.30 A. Meanwhile, the proton of phenolic hydroxylgroup is concertedly shiing to W1 with the length (r3) ofphenolic C–O bond changing to 1.30 A. In IM4-1, the concertedproton transfers have been completely nished, which can becharacterized by the r6 of 1.11 A and distance (r4) of 1.00 Abetween the phenolic hydroxyl hydrogen atom and W1 oxygenatom. The following cleavage of C2–C2a bond is the nal step ofreaction. From IM4-1 to P-1 via TS4-1, the length of C2–C2a bond(r7) is increased from 1.54 A to 3.14 A via 2.32 A. In thiselementary step, the C2a atom gradually becomes to sp3
hybridized.The calculated energy prole of Path-1 is shown in Fig. 10.
The highest point of the energy prole is 28.3 kcal mol�1, whichis a little high for an enzymatic reaction, but this pathway is stillpossible. In addition, the relative energy of the product P-1 ismuch higher than that of IM3, implying the reaction is highlyendothermic.
In Path-2, as shown in Fig. 3, aer the proton transfer from40-NH2 of ThDP to C2a atom to generate IM4-2, the subsequentproton transfer from E121 to 40-NH may occur prior to (Path-2A)or later (Path-2B) than the cleavage of C2–C2a bond. The opti-mized structures of species to generate IM4-2 are shown inFig. 11. In the transition state TS3-2, the distance (r9) betweenthe hydrogen atom of 40-NH2 and C2a atom is 1.32 A and the
Fig. 9 Optimized geometries at the M06-2X/6-31G(d,p) level forvarious species involved in Path-1 of the second half-reaction. The keybond distances are shown in angstrom.
This journal is © The Royal Society of Chemistry 2014
length (r8) of N–H bond has been elongated to 1.41 A. From TS3-2 to IM4-2, r9 further decreased to 1.10 A, indicating thecompletion of proton transfer.
The optimized structures of species for the next two steps,including the proton transfer from E121 to 40-NH and thecleavage of C2–C2a bond, are shown in Fig. 12. In IM4-2, thelength (r7) of C2–C2a bond is 1.55 A and the distance (r11) ofhydrogen bond between the hydrogen atom of E121 carboxylgroup and nitrogen atom of 40-NH is 2.12 A. In Path-2A, theproton transfer occurs rstly to form IM5-2A in which r11changes to 1.02 A from 1.24 A in TS4-2A. Then the C2–C2a bondbroke down to generate P-2A via TS5-2A, and the distance (r7) ofC2–C2a bond changes accordingly from 1.55 A to 3.27 A via 2.27A. In the other pathway Path-2B, from IM4-2, the bond distance(r7) of C2–C2a is rstly increased to 2.28 A in TS4-2B and furtherlengthened to 3.04 A in IM5-2B. Then the following protontransfer occurs to complete the reaction to generate P-2B. Bycomparing the products of the two pathways, some structuraldifferences can be found between P-2A and P-2B, which may bethe main reason leading to different total energies of P-2A andP-2B.
The calculated energy proles of Path-2 are shown in Fig. 13.The rst step to generate IM4-2 corresponds to a high energybarrier of 21.8 kcal mol�1. In the following reactions, both inPath-2A and Path-2B, the energy barriers for the proton trans-fers from E121 to 40-NH are calculated to be�20 kcal mol�1, andthose of C2–C2a bond cleavages are much lower (3.2 and 3.9 kcalmol�1 in Path-2A in Path-2B, respectively). Due to the productsP-2A and P-2B are derived from different pathways, a �4.0 kcalmol�1 energy difference was found. But from energy point ofview, the overall energy barrier of Path-2A is higher than that ofPath-2B, thus the later pathway should be more possible.Moreover, the overall barrier of Path-1 is 6.5 kcal mol�1 higherthat of Path-2B, therefore, Path-2B is the most favorable reac-tion pathway.
In general, based on our calculation results, the OXC-catalyzed reaction was suggested to contain ve elementarysteps as in Path-2B: (1) the ligation of ThDP with the substrate
RSC Adv., 2014, 4, 35777–35788 | 35783

Fig. 11 Optimized geometries at the M06-2X/6-31G(d,p) level for various species involved in the proton transfer from 40-NH2 of ThDP to O2a
atom in Path-2. The key bond distances are shown in angstrom.
Fig. 12 Optimized geometries at the M06-2X/6-31 G(d,p) level for various species involved in the proton transfer from E121 to 40-NH and thecleavage of C2–C2a bond in Path-2A and Path-2B. The key bond distances are shown in angstrom.
Fig. 13 Energy profiles for Path-2A and Path-2B of the second half-reaction in solvent phase (PCM, 3 ¼ 4). The ZPE-corrected relativeenergies (in kcal mol�1) are obtained at the M06-2X/6-311++G(2d,2p)//M06-2X/6-31G(d,p) level.
RSC Advances Paper
oxalyl-CoA; (2) the decarboxylation of a-oxalyl-CoA-ThDP inter-mediate and the liberation of CO2; (3) the proton transfer from40-NH2 to C2a atom; (4) the cleavage of C2–C2a bond to regen-erate formyl-CoA, and (5) the proton transfer from E121 to 40-NH. In this pathway, the proton transfer from 40-NH2 of ThDP tothe C2a atom of the HDC-ThDP intermediate is calculated to bethe rate-limiting step.
35784 | RSC Adv., 2014, 4, 35777–35788
Although Path-2B is themost favorable pathway, if Path-2B isblocked, the reactionmay follow Path-1. However, as mentionedabove, Path-1 corresponds to a higher overall energy barrierthan Path-2B and is highly endothermic, the enzymatic activitymay be reduced at some extent. In the previous mutant stud-ies,18b replacement of some residues was proved to signicantlyreduce but not completely abolish the activity, supporting ourcalculation results. The authors also suggested that no sidechains of the enzyme directly participate in proton transferexcept the glutamic acid (Glu-56).18b However, our calculationsrevealed that both of residues Y483 and E121 are involved in thecatalytic reaction. In Path-1, Y483 donates its proton of phenolichydroxyl group to W1 which donates a proton to C2a atom. InPath-2B, E121 acts as a proton donor in the second half-reaction. Thus, we can conclude that the signicant but notcompletely loss of enzymatic activities of mutants of E121Q,E121A, Y483A and Y483F may be attribute to that, in thesemutants the reaction follow different pathways. In other words,if E121 was mutated, Y483 can act as the proton donor, and viceversa.
3.2 Role of some key residues
Some hydrogen bonds are well kept during the reaction. Thelengths of these hydrogen bonds in all species are given inTables 1 and 2. The water molecule W2 always forms hydrogenbonds with surrounding residues I34, Y120 and E121, and thehydrogen bonds formed by E56 and G426 with ThDP are also
This journal is © The Royal Society of Chemistry 2014

Fig. 14 Optimized geometries of the species and energy profiles ofY120A mutant in the rate-liming steps of Path-1 (a) and Path-2 (b). Thekey bond distances are shown in angstrom. The ZPE-corrected relativeenergies (in kcal mol�1) are obtained at the M06-2X/6-311++G(2d,2p)//M06-2X/6-31G(d,p) level.
Paper RSC Advances
very stable. This indicates their roles in stabilizing the active sitepocket.
For residue Y120, pervious mutant studies showed that itsreplacement by phenylalanine or alanine residues can signi-cantly inuence the activities of the enzyme. Tables 1 and 2show that, in the optimized reactant R the hydrogen bonddistance (d2) between the hydrogen atom of phenolic hydroxylgroup of Y120 and carbonyl oxygen atom of oxalyl-CoA is 1.86 A,which are further decreased in the following intermediates, forexample, they are 1.48 and 1.43 A in IM2 and IM3, respectively.To further explore the role of Y120, we optimized the structuresof mutant Y120A for proton transfers from W1 or 40-NH2 to C2a
atom in Path-1 and Path-2, and the corresponding energyproles are shown in Fig. 14. Aer mutation of Y120, the energybarriers for the proton transfers from W1 or 40-NH2 to C2a atomare both largely increased (23.6 and 28.5 kcal mol�1 for Path-1and Path-2, respectively). These calculation results imply thatthe loss of hydrogen bond between Y120 and substrate cangreatly raise the energy barriers of the reaction. Thus, theresidue Y120 plays an important role in stabilizing the hydroxylanion during the reaction.
4. Conclusion
In this paper, the catalytic mechanism of OXC has been inves-tigated by using DFT method with cluster models. Based on ourcalculations, the most possible reaction pathway, the detail ofeach elementary step, and the roles of some key residues havebeen determined. Our calculation results indicate that the rst-half reaction, which includes the ligation of ThDP with thesubstrate oxalyl-CoA and the decarboxylation of a-oxalyl-CoA-ThDP intermediate and the liberation of CO2, can proceedsrapidly, which agrees well with the experimental observation. Inthe second half-reaction which contains the protonation ofHDC-ThDP intermediate and the cleavage of its C2–C2a bond,the 4-NH2 of ThDP is suggested to be the proton donor, whichabstracts a proton from the nearby residue E121. At the M06-2X/6–311++G(2d,2p)//M06-2X/6–31G(d,p) level of theory, the rate-limiting step is calculated to be the proton transfer from 40-NH2 to the C2a atom of HDC-ThDP intermediate with an energybarrier of 21.8 kcal mol�1. However, if this pathway is blockedby mutating some residues, the reaction may follow the mech-anism as Path-1, in which Y483 acts as the proton donor and
This journal is © The Royal Society of Chemistry 2014
uses a water molecule as mediator. These ndings can explainthe experimental observation that replacement of residues Y483or E121 signicantly reduces but does not completely abolishthe activity of enzyme. During the reaction, in the intermediateswith the C2a atom in sp3 hybridization, the O2a always exists inits hydroxyl anion form and forms hydrogen bonds with residueY120 and/or 40-NH2 of ThDP. Besides, water moleculeW2 alwaysforms hydrogen bonds with the surrounding residues I34, Y120and E121, and residues E56 and G426 always form stablehydrogen bonds with ThDP, which plays important roles instabilizing the intermediates and transition states. Our resultsmay provide useful information for exploring the enzymaticmechanism and developing biocatalytic applications on treat-ing the oxalate-related diseases.
Acknowledgements
This work was supported by the National Natural ScienceFoundation of China (21173129, 21373125, 31200048).
References
1 (a) H. E. Williams and L. H. Smith Jr, Disorders of oxalatemetabolism, Am. J. Med., 1968, 45, 715–735; (b)M. M. Rahman, R. B. Abdullah and W. E. Wan Khadijah, Areview of oxalate poisoning in domestic animals: toleranceand performance aspects, J. Anim. Physiol. Anim. Nutr.,2013, 97, 605–614.
2 (a) F. L. Coe, J. H. Parks and J. R. Asplin, The pathogenesisand treatment of kidney stones, N. Engl. J. Med., 1992, 327,1141–1152; (b) A. Hesse and R. Siener, Current aspects ofepidemiology and nutrition in urinary stone disease, WorldJ. Urol., 1997, 15, 165–171.
3 E. M. Worcester, Y. Nakagawa, D. A. Bushinsky and F. L. Coe,Evidence that serum calcium oxalate supersaturation is aconsequence of oxalate retention in patients with chronicrenal failure, J. Clin. Invest., 1986, 77, 1888–1896.
4 J. W. O'Callaghan, S. M. Arbuckle, P. W. Craswell, P. P. Boyle,J. W. Searle andW. R. Smythe, Rapid progression of oxalosis-induced cardiomyopathy despite adequate haemodialysis,Miner. Electrolyte Metab., 1984, 10, 48–51.
5 R. A. Rodby, T. S. Tyszka and J. W. Williams, Reversal ofcardiac dysfunction secondary to type 1 primaryhyperoxaluria aer combined liver-kidney transplantation,Am. J. Med., 1991, 90, 498–504.
6 M. Hatch, R. W. Freel and N. D. Vaziri, Mechanisms ofoxalate absorption and secretion across the rabbit distalcolon, Puegers Arch., 1994, 426, 101–109.
7 (a) J. Jiang, J. Knight, L. H. Easter, R. Neiberg, R. P. Holmesand D. G. Assimos, Impact of dietary calcium and oxalate,and Oxalobacter formigenes colonization on urinary oxalateexcretion, J. Urol., 2011, 186, 135–139; (b) J. N. Lange,K. D. Wood, P. W. Mufarrij, M. F. Callahan, L. Easter,J. Knight, R. P. Holmes and D. G. Assimos, The impact ofdietary calcium and oxalate ratios on stone risk, Urology,2012, 79, 1226–1229.
RSC Adv., 2014, 4, 35777–35788 | 35785

RSC Advances Paper
8 (a) O. Ivanovski and T. B. Drueke, A new era in the treatmentof calcium oxalate stones?, Kidney Int., 2013, 83, 998–1000;(b) R. P. Holmes, H. O. Goodman and D. G. Assimos,Contribution of dietary oxalate to urinary oxalate excretion,Kidney Int., 2001, 59, 270–276.
9 (a) D. W. Kaufman, J. P. Kelly, G. C. Curhan, T. E. Anderson,S. P. Dretler, G. M. Preminger and D. R. Cave, Oxalobacterformigenes may reduce the risk of calcium oxalate kidneystones, J. Am. Soc. Nephrol., 2008, 19, 1197–1203; (b)R. Siener, U. Bangen, H. Sidhu, R. Honow, G. von Unruhand A. Hesse, The role of Oxalobacter formigenescolonization in calcium oxalate stone disease, Kidney Int.,2013, 83, 1144–1149; (c) J. Knight, R. Deora, D. G. Assimosand R. P. Holmes, The genetic composition of Oxalobacterformigenes and its relationship to colonization and calciumoxalate stone disease, Urolithiasis, 2013, 41, 187–196; (d)C. S. Stewart, S. H. Duncan and D. R. Cave, Oxalobacterformigenes and its role in oxalate metabolism in the humangut, FEMS Microbiol. Lett., 2004, 230, 1–7.
10 M. J. Allison, K. A. Dawson, W. R. Mayberry and J. G. Foss,Oxalobacter formigenes gen. nov., sp. nov.: oxalate-degrading anaerobes that inhabit the gastrointestinal tract,Arch. Microbiol., 1985, 141, 1–7.
11 (a) M. Hatch, J. Cornelius, M. Allison, H. Sidhu, A. Peck andR. W. Freel, Oxalobacter sp. reduces urinary oxalateexcretion by promoting enteric oxalate secretion, KidneyInt., 2006, 69, 691–698; (b) R. Kumar, M. Mukherjee,M. Bhandari, A. Kumar, H. Sidhu and R. D. Mittal, Role ofOxalobacter formigenes in calcium oxalate stone disease: astudy from North India, Eur. Urol., 2002, 41, 318–322; (c)C. Kwak, H. K. Kim, E. C. Kim, M. S. Choi and H. H. Kim,Urinary oxalate levels and the enteric bacteriumOxalobacter formigenes in patients with calcium oxalateurolithiasis, Eur. Urol., 2003, 44, 475–481.
12 (a) S. A. Troxel, H. Sidhu, P. Kaul and R. K. Low, IntestinalOxalobacter formigenes colonization in calcium oxalatestone formers and its relation to urinary oxalate, J.Endourol., 2003, 17, 173–176; (b) B. Hoppe, J. W. Groothoff,S. A. Hulton, P. Cochat, P. Niaudet, M. J. Kemper,G. Deschenes, R. Unwin and D. Milliner, Efficacy andsafety of Oxalobacter formigenes to reduce urinary oxalatein primary hyperoxaluria, Nephrol., Dial., Transplant., 2011,26, 3609–3615; (c) R. Kumar, U. C. Ghoshal, G. Singh andR. D. Mittal, Infrequency of colonization with Oxalobacterformigenes in inammatory bowel disease: possible role inrenal stone formation, J. Gastroenterol. Hepatol., 2004, 19,1403–1409.
13 (a) E. Leumann, B. Hoppe, T. Neuhaus and N. Blau, Efficacyof oral citrate administration in primary hyperoxaluria,Nephrol., Dial., Transplant., 1995, 10, 14–16; (b)D. S. Milliner, J. T. Eickholt, E. J. Bergstralh, D. M. Wilsonand L. H. Smith, Results of long term treatment withorthophosphate and pyridoxine in patients with primaryhyperoxaluria, N. Engl. J. Med., 1994, 331, 1553–1558; (c)B. Hoppe, K. Latta, C. von Schnakenburg andM. J. Kemper, on behalf of the Arbeitsgemeinscha fur
35786 | RSC Adv., 2014, 4, 35777–35788
padiatrische Nephrologie. Primary Hyperoxaluria: theGerman experience, Am. J. Nephrol., 2005, 25, 276–281.
14 (a) B. Hoppe, B. Beck, N. Gatter, G. von Unruh, A. Tischer,A. Hesse, N. Laube, P. Kaul and H. Sidhu, Oxalobacterformigenes: a potential tool for the treatment of primaryhyperoxaluria type 1, Kidney Int., 2006, 70, 1305–1311; (b)H. Sidhu, M. E. Schmidt, J. G. Cornelius, S. Thamilselvan,S. R. Khan, A. Hesse and A. B. Peck, Direct correlationbetween hyperoxaluria/oxalate stone disease and theabsence of the gastrointestinal tract-dwelling bacteriumOxalobacter formigenes: possible prevention by gutrecolonization or enzyme replacement therapy, J. Am. Soc.Nephrol., 1999, 10, S334–S340; (c) B. Hoppe, K. Dittlich,H. Fehrenbach, G. Plum and B. B. Beck, Reduction ofplasma oxalate levels by oral application of Oxalobacterformigenes in 2 patients with infantile oxalosis, Am. J.Kidney Dis., 2011, 58, 453–455; (d) D. Grujic, E. C. Salido,B. C. Shenoy, C. B. Langman, M. E. McGrath, R. J. Patel,A. Rashid, S. Mandapati, C. W. Jung and A. L. Margolin,Hyperoxaluria is reduced and nephrocalcinosis preventedwith an oxalate-degrading enzyme in mice withhyperoxaluria, Am. J. Nephrol., 2008, 29, 86–93; (e)D. Milliner, Treatment of the primary hyperoxalurias: anew chapter, Kidney Int., 2006, 70, 1198–2000; (f) S. Siva,E. R. Barrack, G. Reddy, V. Thamilselvan, S. Thamilselvan,M. Menon and M. Bhandari, A critical analysis of the roleof gut Oxalobacter formigenes in oxalate stone disease, BJUInt., 2009, 103, 18–21.
15 (a) S. H. Duncan, A. J. Richardson, P. Kaul, R. P. Holmes,M. J. Allison and C. S. Stewart, Oxalobacter formigenes andits potential role in human health, Appl. Environ.Microbiol., 2002, 68, 3841–3847; (b) H. Sidhu, M. J. Allison,J. M. Chow, A. Clark and A. B. Peck, Rapid reversal ofhyperoxaluria in a rat model aer probiotic administrationof Oxalobacter formigenes, J. Urol., 2001, 166, 1487–1491.
16 (a) R. D. Mittal and R. Kumar, Gut-inhabiting bacteriumOxalobacter formigenes: role in calcium oxalate urolithiasis,J. Endourol., 2004, 18, 418–424; (b) R. D. Mittal, R. Kumar,B. Mittal, R. Prasad and M. Bhandari, Stone composition,metabolic prole and the presence of the gut-inhabitingbacterium Oxalobacter formigenes as risk factors for renalstone formation, Med. Princ. Pract., 2003, 12, 208–213.
17 (a) S. Ricagno, S. Jonsson, N. Richards and Y. Lindqvist,Formyl-CoA transferase encloses the CoA binding site atthe interface of an interlocked dimer, EMBO J., 2003, 22,3210–3219; (b) S. Jonsson, S. Ricagno, Y. Lindqvist andN. G. J. Richards, Kinetic and mechanistic characterizationof the formyl-CoA transferase from Oxalobacter formigenes,J. Biol. Chem., 2004, 279, 36003–36012; (c) C. L. Berthold,C. G. Toyota, N. G. Richards and Y. Lindqvist,Reinvestigation of the catalytic mechanism of formyl-CoAtransferase, a class III CoA-transferase, J. Biol. Chem., 2008,283, 6519–6529.
18 (a) C. L. Berthold, P. Moussatche, N. G. Richards andY. Lindqvist, Structural basis for activation of the thiamindiphosphate-dependent enzyme oxalyl-CoA decarboxylaseby adenosine diphosphate, J. Biol. Chem., 2005, 280,
This journal is © The Royal Society of Chemistry 2014

Paper RSC Advances
41645–41654; (b) C. L. Berthold, C. G. Toyota, P. Moussatche,M. D. Wood, F. Leeper, N. G. Richards and Y. Lindqvist,Crystallographic snapshots of oxalyl-CoA decarboxylasegive insights into catalysis by nonoxidative ThDP-dependent decarboxylases, Structure, 2007, 15, 853–861; (c)T. Werther, A. Zimmer, G. Wille, R. Golbik, M. S. Weissand S. Konig, New insights into structure–functionrelationships of oxalyl CoA decarboxylase from Escherichiacoli, FEBS J., 2010, 277, 2628–2640.
19 (a) Y. A. Muller, Y. Lindqvist, W. Furey, G. E. Schulz,F. Jordan and G. Schneider, A thiamin diphosphatebinding fold revealed by comparison of the crystalstructures of transketolase, pyruvate oxidase and pyruvatedecarboxylase, Structure, 1993, 1, 95–103; (b) R. A. Frank,F. J. Leeper and B. F. Luisi, Structure, mechanism andcatalytic duality of thiamine-dependent enzymes, Cell. Mol.Life Sci., 2007, 64, 892–905.
20 B. Shaanan and D. M. Chipman, Reaction mechanism ofthiamin diphosphate enzymes: New insights into the roleof a conserved glutamate residue, FEBS J., 2009, 276, 2447–2453.
21 (a) R. B. Wu, S. L. Wang, N. J. Zhou, Z. X. Cao andY. K. Zhang, A proton-shuttle reaction mechanism forhistone deacetylase 8 and the catalytic role of metal ions, J.Am. Chem. Soc., 2010, 132, 9471–9479; (b) D. G. Xu, H. Guoand Q. Cui, Antibiotic deactivation by a dizinc beta-lactamase: mechanistic insights from QM/MM and DFTstudies, J. Am. Chem. Soc., 2007, 129, 10814–10822; (c)D. G. Xu and H. Guo, Quantum mechanical/molecularmechanical and density functional theory studies of aprototypical zinc peptidase (carboxypeptidase A) suggest ageneral acid–general base mechanism, J. Am. Chem. Soc.,2009, 131, 9780–9788.
22 (a) M. Fries, H. I. Jung and R. N. Perham, Reactionmechanism of the heterotetrameric (a2b2) E1 componentof 2-oxo acid dehydrogenase multienzyme complexes,Biochemistry, 2003, 42, 6996–7002; (b) W. Versees,S. Spaepen, M. D. Wood, F. J. Leeper, J. Vanderleyden andJ. Steyaert, Molecular mechanism of allosteric substrateactivation in a thiamine diphosphate-dependentdecarboxylase, J. Biol. Chem., 2007, 282, 35269–35278; (c)L. Mitschke, C. Parthier, K. Schroder-Tittmann, J. Coy,S. Ludtke and K. Tittmann, The crystal structure of humantransketolase and new insights into its mode of action, J.Biol. Chem., 2010, 285, 31559–31570.
23 (a) J. Y. Wang, H. Dong, S. H. Li and H. W. He, Theoreticalstudy toward understanding the catalytic mechanism ofpyruvate decarboxylase, J. Phys. Chem. B, 2005, 109, 18664–18672; (b) Q. Q. Hou, J. Gao, Y. J. Liu and C. B. Liu, A QM/MM study on the catalytic mechanism of pyruvatedecarboxylase, Theor. Chem. Acc., 2012, 131, 1280; (c)R. Friedemann, K. Tittmann, R. Golbik and G. Hubner,DFT and MP2 studies on the C2–C2 bond cleavage inthiamin catalysis, J. Mol. Catal. B: Enzym., 2009, 61, 36–38.
24 (a) G. Jana, V. Jimenez, J. Villa-Freixa, X. Prat-Resina,E. Delgado and J. B. Alderete, Computational study on thecarboligation reaction of acetohydroxyacid synthase: New
This journal is © The Royal Society of Chemistry 2014
approach on the role of the HEThDP-intermediate,Proteins, 2010, 78, 1774–1788; (b) Y. Xiong, J. Liu,G. F. Yang and C. G. Zhan, Computational determinationof fundamental pathway and activation barriers foracetohydroxyacid synthase-catalyzed condensationreactions of a-keto acids, J. Comput. Chem., 2010, 31, 1592–1602.
25 R. S. Assary and L. J. Broadbelt, Computatinal screening ofnovel thiamine-catalyzed decarboxylation reactions of 2-keto acids, Bioprocess Biosyst. Eng., 2011, 34, 375–388.
26 (a) C. Acosta-Silva, J. Bertran, V. Branchadell and A. Oliva,Quantum-mechanical study on the mechanism of peptidebond formation in the ribosome, J. Am. Chem. Soc., 2012,134, 5817–5831; (b) M. E. S. Lind and F. Himo, Quantumchemistry as a tool in asymmetric biocatalysis: Limoneneepoxide hydrolase test case, Angew. Chem., Int. Ed., 2013,52, 4563–4567; (c) A. Miłaczewska, E. Broclawik andT. Borowski, On the catalytic mechanism of (S)-2-hydroxypropylphosphonic acid epoxidase (HppE): A hybridDFT study, Chem.–Eur. J., 2013, 19, 771–781; (d) T. Marino,N. Russo and M. Toscano, Catalytic mechanism of thearylsulfatase promiscuous enzyme from Pseudomonasaeruginosa, Chem.–Eur. J., 2013, 19, 2185–2192; (e) F. Himoand P. E. M. Siegbahn, Recent developments of thequantum chemical cluster approach for modeling enzymereactions, JBIC, J. Biol. Inorg. Chem., 2009, 14, 643–651; (f)M. R. A. Blomberg and P. E. M. Siegbahn, Quantumchemistry as a tool in bioenergetics, Biochim. Biophys. Acta,2010, 1797, 129–142; (g) O. Amata, T. Marino, N. Russoand M. Toscano, A proposal for mitochondrial processingpeptidase catalytic mechanism, J. Am. Chem. Soc., 2011,133, 17824–17831; (h) E. A. C. Bushnell, R. Gherib andJ. W. Gauld, Insights into the catalytic mechanism of coralallene oxide synthase: A dispersion corrected densityfunctional theory study, J. Phys. Chem. B, 2013, 117, 6701–6710; (i) J. J. Robinet, K.-B. Cho and J. W. Gauld, A densityfunctional theory investigation on the mechanism of thesecond half-reaction of nitric oxide synthase, J. Am. Chem.Soc., 2008, 130, 3328–3334.
27 (a) A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu andM. H. Olsson, Electrostatic basis for enzyme catalysis,Chem. Rev., 2006, 106, 3210–3235; (b) T. Kamachi,T. Nakayama, O. Shitamichi, K. Jitsumori, T. Kurihara,N. Esaki and K. Yoshizawa, The catalytic mechanism ofuoroacetate dehalogenase: A computational explorationof biological dehalogenation, Chem.–Eur. J., 2009, 15,7394–7403; (c) X. Jun, J. Z. H. Zhang and Y. Xiang, Abinitio QM/MM free energy simulations of peptide bondformation in the ribosome support an eight-memberedring reaction mechanism, J. Am. Chem. Soc., 2012, 134,16424–16429; (d) V. Lopez-Canut, J. J. Ruiz-Pernıa,R. Castillo, V. Moliner and I. Tunon, Hydrolysis ofphosphotriesters: A theoretical analysis of the enzymaticand solution mechanisms, Chem.–Eur. J., 2012, 18, 9612–9621; (e) C. Li and S. Shaik, How do peruorinatedalkanoic acids elicit cytochrome P450 to catalyze methane
RSC Adv., 2014, 4, 35777–35788 | 35787

RSC Advances Paper
hydroxylation? An MD and QM/MM study, RSC Adv., 2013, 3,2995–3005.
28 (a) R. Sevastik and F. Himo, Reaction mechanism of cis-3-chloroacrylic acid dehalogenase: a theoretical study,Bioorg. Chem., 2007, 35, 444–457; (b) K. H. Hopmann andF. Himo, Quantum chemical modeling of thedehalogenation reaction of haloalcohol dehalogenase, J.Chem. Theory Comput., 2008, 4, 1129–1137; (c) P. Georgievaand F. Himo, Quantum chemical modeling of enzymaticreactions: the case of histone lysine methyltransferase, J.Comput. Chem., 2010, 31, 1707–1714; (d) R. Z. Liao,P. Georgieva, J. G. Yu and F. Himo, Mechanism of mycolicacid cyclopropane synthase: a theoretical study,Biochemistry, 2011, 50, 1505–1513.
29 (a) M. H. M. Olsson, C. R. Søndergard, M. Rostkowski andJ. H. Jensen, PROPKA3: consistent treatment of internaland surface residues in empirical pKa predictions, J. Chem.Theory Comput., 2011, 2, 525–537; (b) C. R. Søndergaard,M. H. M. Olsson, M. Rostkowski and J. H. Jensen,Improved treatment of ligands and coupling effects inempirical calculation and rationalization of pKa values, J.Chem. Theory Comput., 2011, 7, 2284–2295.
30 (a) J. Y. Wang and S. H. Li, Theoretical study towardunderstanding the catalytic mechanism of pyruvatedehydrogenase multienzyme complex E1 component, J.Theor. Comput. Chem., 2006, 5, 447–459; (b) D. Kern,G. Kern, H. Neef, K. Tittmann, M. Killenberg-Jabs,C. Wikner, G. Schneider and G. Hubner, How thiaminediphosphate is activated in enzymes, Science, 1997, 275,67–70; (c) K. Tittmann, K. Mesch, M. Pohl and G. Hubner,Activation of thiamine diphosphate in pyruvatedecarboxylase from Zymomonas mobilis, FEBS Lett., 1998,441, 404–406; (d) A. Bar-Ilan, V. Balan, K. Tittmann,R. Golbik, M. Vyazmensky, G. Hubner, Z. Barak andD. M. Chipman, Binding and activation of thiamindiphosphate in acetohydroxyacid synthase, Biochemistry,2001, 40, 11946–11954.
31 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria,M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone,B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato,X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng,J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota,R. Fukuda, J. mHasegawa, M. Ishida, T. Nakajima,Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. MontgomeryJr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd,E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith,R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell,J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega,J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken,C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann,
35788 | RSC Adv., 2014, 4, 35777–35788
O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli,J. W. Ochterski, R. L. Martin, K. Morokuma,V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg,S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman,J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09,Revision B.01, Gaussian, Inc., Wallingford, CT, 2010.
32 Y. Zhao and D. G. Truhlar, The M06 suite of densityfunctionals for main group thermochemistry,thermochemical kinetics, noncovalent interactions, excitedstates, and transition elements: Two new functionals andsystematic testing of four M06-class functionals and 12other functional, Theor. Chem. Acc., 2006, 120, 215.
33 (a) V. Barone, M. Cossi and J. Tomasi, Geometryoptimization of molecular structures in solution by thepolarizable continuum model, J. Comput. Chem., 1998, 19,404–417; (b) J. Tomasi and M. Persico, Molecularinteractions in solution: An overview of methods based oncontinuous distributions of the solvent, Chem. Rev., 1994,94, 2027–2094.
34 (a) J. J. Robinet, K. B. Cho and J. W. Gauld, A densityfunctional theory investigation on the mechanism of thesecond half-reaction of nitric oxide synthase, J. Am. Chem.Soc., 2008, 130, 3328–3334; (b) R. J. Liao, W. J. Ding,J. G. Yu, W. H. Fang and R. Z. Liu, Theoretical studies onpyridoxal 50-phosphate-dependent transamination of a-amino acids, J. Comput. Chem., 2008, 29, 1919–1929; (c)P. E. M. Siegbahn and T. Borowski, Modeling enzymaticreactions involving transition metals, Acc. Chem. Res., 2006,39, 729–738; (d) F. Himo, Quantum chemical modeling ofenzyme active sites and reaction mechanisms, Theor.Chem. Acc., 2006, 116, 232–240; (e) M. J. Ramos andP. A. Fernandes, Computational enzymatic catalysis, Acc.Chem. Res., 2008, 41, 689–698.
35 (a) C. Gonzalez and H. B. Schlegel, An improved algorithmfor reaction path following, J. Chem. Phys., 1989, 90, 2154–2161; (b) C. Gonzalez and H. B. Schlegel, Reaction pathfollowing in mass weighted internal coordinates, J. Phys.Chem., 1990, 94, 5523–5527.
36 (a) Y. A. Muller, Y. Lindqvist, W. Furey, G. E. Schuza,F. Jordan and G. Schneider, A thiamin diphosphatebinding old revealed by comparison of the crystalstructures of transketolase pyruvate oxidase and pyruvatedecarboxylase, Structure, 1993, 1, 95–103; (b) Y. A. Mullerand G. E. Schulz, Structure of the thiamine- and avin-dependent enzyme pyruvate oxidase, Science, 1993, 259,965–967.
37 F. Jordan and N. S. Nemeria, Experimental observation ofthiamin diphosphate-bound intermediates on enzymesand mechanistic information derived from theseobservations, Bioorg. Chem., 2005, 33, 190–215.
This journal is © The Royal Society of Chemistry 2014