a pediatrician s practical guide to diagnosing and treating hereditary spherocytosis ... ·...
TRANSCRIPT

A Pediatrician’s Practical Guide toDiagnosing and Treating HereditarySpherocytosis in NeonatesRobert D. Christensen, MDa,b, Hassan M. Yaish, MDb, Patrick G. Gallagher, MDc
abstractNewborn infants who have hereditary spherocytosis (HS) can develop anemiaand hyperbilirubinemia. Bilirubin-induced neurologic dysfunction is less likelyin these neonates if the diagnosis of HS is recognized and appropriatetreatment provided. Among neonates listed in the USA Kernicterus Registry,HS was the third most common underlying hemolytic condition after glucose-6-phosphate dehydrogenase deficiency and ABO hemolytic disease. HS is theleading cause of direct antiglobulin test (direct Coombs) negative hemolyticanemia requiring erythrocyte transfusion in the first months of life. Weanticipate that as physicians become more familiar with diagnosing HS in thenewborn period, fewer neonates with HS will develop hazardoushyperbilirubinemia or present to emergency departments with unanticipatedsymptomatic anemia. We predict that early suspicion, prompt diagnosis andtreatment, and anticipatory guidance will prevent adverse outcomes inneonates with HS. The purpose of this article was to review the neonatalpresentation of HS and to provide practical and up-to-date means ofdiagnosing and treating HS in neonates.
Hereditary spherocytosis (HS) isa heterogeneous disorder in whichabnormalities of red blood cellstructural proteins lead to loss oferythrocyte membrane surface area,resulting in spherical-shaped,hyperdense, poorly deformable redblood cells (Fig 1) with a shortenedlife span.1–5 HS occurs worldwide andaffects individuals from all racial andethnic groups. Individual pediatriciansencounter HS uncommonly, buthospitals and health care systemswith large delivery services regularlydeal with this condition, particularlyin white neonates of northernEuropean ancestry, in whom thecondition can be as frequent as 1 in1000 to 2000 births. Early suspicionand diagnosis of HS allow appropriatemanagement, including provision ofanticipatory guidance to parents,which can reduce the risk of adverseoutcomes.
The purpose of the present article wasto review the neonatal presentationof HS and to provide practical andup-to-date means of diagnosing andtreating HS in neonates.
PATHOGENESIS
The loss of membrane surface area inHS erythrocytes is due to defects invarious erythrocyte membraneproteins, ankyrin-1, band 3, b-spectrin,a-spectrin, and protein 4.2 (Table 1,Fig 2).1–5 Numerous mutations in thegenes encoding these membraneproteins have been described.Destruction of poorly deformable HSerythrocytes in the spleen is theprimary cause of hemolysis in patientswith HS.
The clinical spectrum of HS during theperinatal period ranges from severefetal anemia with hydrops fetalis to theasymptomatic neonate. This wide
Divisions of aNeonatology and bHematology/Oncology,Department of Pediatrics, University of Utah School ofMedicine, Primary Children’s Hospital, IntermountainHealthcare, Salt Lake City, Utah; and cDivision ofNeonatology, Department of Pediatrics, Yale UniversitySchool of Medicine, Yale–New Haven Children’s Hospital,New Haven, Connecticut
Dr Christensen conceptualized and drafted theinitial manuscript; Drs Yaish and Gallagher authoredthe manuscript; and all authors approved the finalversion as submitted.
www.pediatrics.org/cgi/doi/10.1542/peds.2014-3516
DOI: 10.1542/peds.2014-3516
Accepted for publication Jan 29, 2015
Address correspondence to Robert D. Christensen,MD, Department of Pediatrics, Room 2N131, 295Chipeta Way, University of Utah, Salt Lake City, UT84108. E-mail: [email protected]
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online,1098-4275).
Copyright © 2015 by the American Academy ofPediatrics
FINANCIAL DISCLOSURE: The authors have indicatedthey have no financial relationships relevant to thisarticle to disclose.
FUNDING: This work was supported by NIH grantRO1-HL65448. Funded by the National Institutes ofHealth.
POTENTIAL CONFLICT OF INTEREST: The authors haveindicated they have no potential conflicts of interestto disclose.
PEDIATRICS Volume 135, number 6, June 2015 STATE-OF-THE-ART REVIEW ARTICLEby guest on August 17, 2016Downloaded from

range is due, in part, to the variousgenes and specific mutationsinvolved, as well as to the presence orabsence of coinherited conditions. Forinstance, HS coinherited withmutations or polymorphisms of genesinvolved in bilirubin uptake intohepatocytes (SLC01B1) orintrahepatic bilirubin conjugation(UGT1A1) can increase the risk ofhazardous hyperbilirubinemia andkernicterus.6–9
MAKING THE DIAGNOSIS OF HS INA NEONATE
As with any relatively uncommonneonatal disorder, the first step inmaking the diagnosis of HS isconsidering it in the differentialdiagnosis. Thus, a basicunderstanding of HS is useful tophysicians caring for neonates. Wehave found that the triad of anemia,splenomegaly, and jaundice, which isfound in older children and adultswith HS, is rare in neonates. Morethan one-half of neonates with HS are
not anemic in the first week of life,and splenomegaly is rarelydetected.10 Jaundice is the mostcommon presenting feature of HS inneonates.11 In addition, the typicallysluggish erythropoietic response ofneonates often renders thereticulocyte count low relative to thedegree of anemia10; spherocytes areless often observed on the bloodsmear of neonates; and other markersof hemolysis seen in older patients,such as low haptoglobin levels,12 maybe poor indicators of hemolysis in theneonate.
Severe neonatal jaundice can be theresult of various underlyingcauses.13–16 Unfortunately, asillustrated by Johnson et al,13 evenwhen hyperbilirubinemia is severeenough to cause kernicterus, theunderlying cause of the jaundicefrequently remains unidentified.Perhaps the intense focus on reducingthe elevated bilirubin level intoa safe range sometimes results inneglecting evaluation of how the
bilirubin became so high in the firstplace.14
Approximately 65% of neonates withHS have a parent with HS.1,3,5 Whena parent has HS, it is important thatthis information be placedprominently in the prenatal recordand communicated verbally, beforebirth, to the physicians and thehospital staff who will be providingneonatal care. All parents with HSshould be encouraged tocommunicate this information totheir infant’s physician beforedelivery. Failure to do so sometimesoccurs when the affected parent hasbeen asymptomatic since undergoingsplenectomy as a child and has all butforgotten about the condition, andfails to consider that it might beproblematic for the newborn infant.It can be helpful to specificallyinquire of parents of anemic and/orjaundiced neonates about a familyhistory of anemia, jaundice,splenectomy, or early gallstones.
One way to suspect HS in a jaundicedneonate is to obtain a completeblood cell count for interpretation ofthe red blood cell indices, inparticular the mean corpuscularhemoglobin concentration (MCHC)and the mean corpuscular volume(MCV), and to examine the peripheralblood smear for the presence ofspherocytes and polychromasia.17
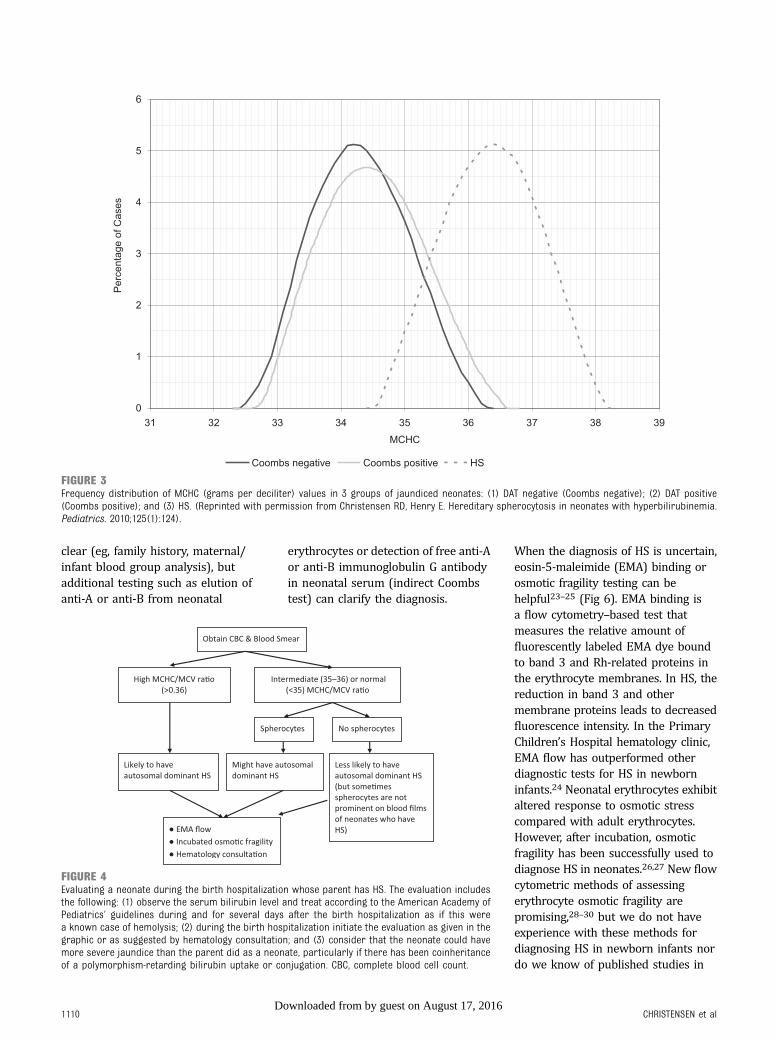
Typically, a neonate with HS will havean elevated MCHC. Figure 3 displaysthe histograms of MCHCmeasurements in 3 groups ofjaundiced neonates: (1) directantiglobulin test (DAT)-negativejaundice; (2) DAT-positive jaundice(primarily ABO hemolytic disease);and (3) HS. The histogram revealssome overlap in MCHC, but, as
TABLE 1 Erythrocyte Membrane Proteins Involved in HS
Protein Gene Chromosomal Location Percentage of HS Cases Typical Severity Inheritance
Ankyrin-1 ANK1 8p11.2 40–50 Mild to moderate Autosomal dominantBand 3 SLC4A1 17q21 20–35 Mild to moderate Autosomal dominantb-spectrin SPTB 14q23-24.1 15–30 Mild to moderate Autosomal dominanta-spectrin SPTA1 1q22-23 ,5 Severe Autosomal recessiveProtein 4.2 EPB42 15q15-21 ,5 Mild to moderate Autosomal recessive
FIGURE 1Photomicrograph of a stained blood film from a neonate with hereditary spherocytosis (band 3mutation). Several spherocytes are present in this field, along with polychromatophilic erythrocytesand 2 nucleated red blood cells.
1108 CHRISTENSEN et alby guest on August 17, 2016Downloaded from

a general rule, if a neonate’s MCHC iselevated (.36.5 to 37 g/dL), HS islikely.11 Another way to estimatewhether a neonate has HS takesadvantage of the fact that in mostneonates with HS, the MCV is low.Based on this scenario, the neonatalHS ratio can be calculated by dividingthe MCHC by the MCV. Using thisratio, little overlap is seen betweennormal neonates and those laterproven to have HS. In fact, in theIntermountain Healthcare database,a neonatal HS ratio .0.36 indicatesthat HS is present with 97%sensitivity, .99% specificity, and.99% negative predictive value.18
It is likely that different makes andmodels of automated cell countersgenerate slight differences inerythrocyte indices, such as the MCVand MCHC. Validation of the neonatalHS ratio on all models of counterswould be useful. The meanhemoglobin content of all themeasured red blood cells as well as
reticulocyte parameters cansometimes be helpful in suggestingHS,19 but we are unaware of reportsof their use in newborn infants.
Figure 4 reviews ourrecommendations for how to evaluatea neonate for HS when a parent isknown to have HS. Because thecommon forms of HS are inherited inan autosomal dominant manner, eachchild born to a parent with HS hasa 50% chance of inheriting thedisorder. The key element in thealgorithm is to treat the neonate as ifhe or she has HS until provenotherwise. In addition to obtainingerythrocyte indices, this approachincludes peripheral smearexamination and reticulocyte count,adhering to the American Academy ofPediatrics’ guidelines for bilirubinmonitoring in the birth hospital,aggressive phototherapy whenindicated, and a follow-up bilirubinassessment no later than 24 hoursafter the hospital discharge.20,21
Figure 5 reviews ourrecommendations for evaluatinga jaundiced neonate for HS whenneither parent has HS. There is noneed to wait until the neonate isseveral months old, or untilsignificant anemia develops, to beginthe evaluation. By following thealgorithm in Fig 4 (parent has HS) orFig 5 (parent does not have HS), thediagnosis can often be made duringthe birth hospitalization.
The presence of spherocytes onperipheral blood smear is helpfulwhen considering the diagnosis of HS(Fig 1),14 but up to one-third ofneonates with HS do not havespherocytes identified prominently ontheir blood smear. Some neonates withband 3 deficiency have pincered redblood cells.22 A subset of neonateswith DAT-negative ABOincompatibility severe enough toproduce spherocytes on peripheralsmear have been described. In mostcases, the differentiation from HS is
FIGURE 2Schematic diagram of the erythrocyte membrane. The upper portion represents the erythrocyte’s external surface, separated from the internalstructures by the lipid bilayer. Proteins shown where mutations result in HS include band 3, ankyrin-1, a-spectrin, b-spectrin, and protein 4.2. (Reprintedwith permission from Gallagher PG. Abnormalities of the erythrocyte membrane. Pediatr Clin North Am. 2013;60(6):1358).
PEDIATRICS Volume 135, number 6, June 2015 1109by guest on August 17, 2016Downloaded from
clear (eg, family history, maternal/infant blood group analysis), butadditional testing such as elution ofanti-A or anti-B from neonatal
erythrocytes or detection of free anti-Aor anti-B immunoglobulin G antibodyin neonatal serum (indirect Coombstest) can clarify the diagnosis.
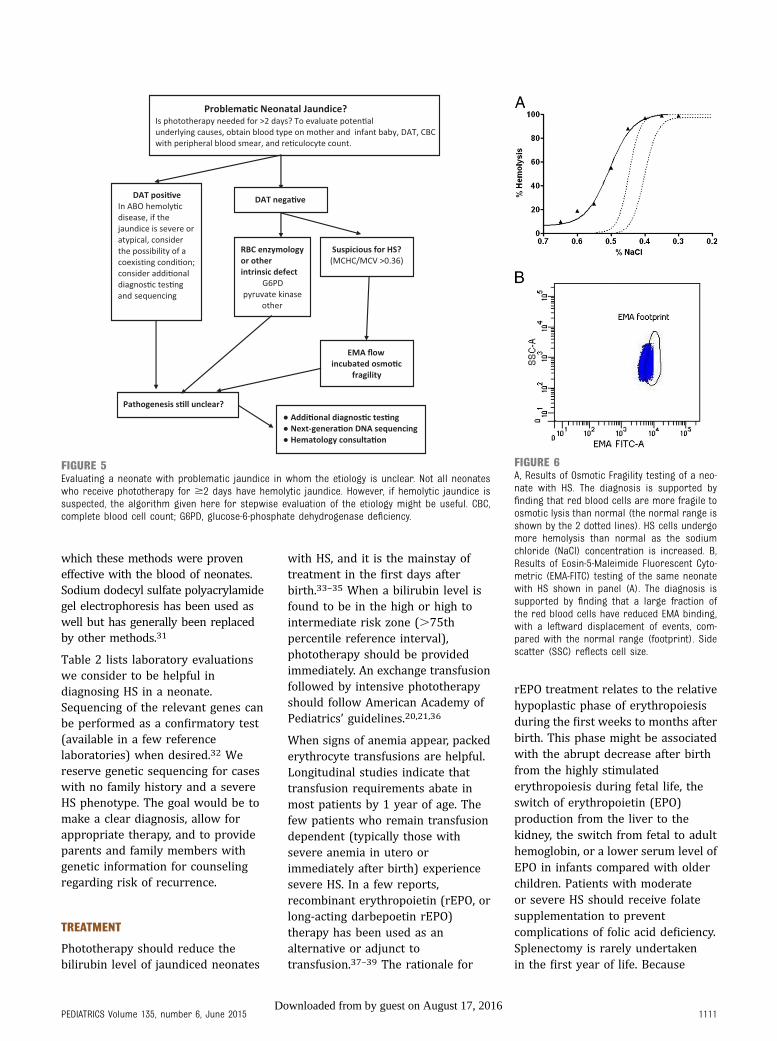
When the diagnosis of HS is uncertain,eosin-5-maleimide (EMA) binding orosmotic fragility testing can behelpful23–25 (Fig 6). EMA binding isa flow cytometry–based test thatmeasures the relative amount offluorescently labeled EMA dye boundto band 3 and Rh-related proteins inthe erythrocyte membranes. In HS, thereduction in band 3 and othermembrane proteins leads to decreasedfluorescence intensity. In the PrimaryChildren’s Hospital hematology clinic,EMA flow has outperformed otherdiagnostic tests for HS in newborninfants.24 Neonatal erythrocytes exhibitaltered response to osmotic stresscompared with adult erythrocytes.However, after incubation, osmoticfragility has been successfully used todiagnose HS in neonates.26,27 New flowcytometric methods of assessingerythrocyte osmotic fragility arepromising,28–30 but we do not haveexperience with these methods fordiagnosing HS in newborn infants nordo we know of published studies in
FIGURE 3Frequency distribution of MCHC (grams per deciliter) values in 3 groups of jaundiced neonates: (1) DAT negative (Coombs negative); (2) DAT positive(Coombs positive); and (3) HS. (Reprinted with permission from Christensen RD, Henry E. Hereditary spherocytosis in neonates with hyperbilirubinemia.Pediatrics. 2010;125(1):124).
FIGURE 4Evaluating a neonate during the birth hospitalization whose parent has HS. The evaluation includesthe following: (1) observe the serum bilirubin level and treat according to the American Academy ofPediatrics’ guidelines during and for several days after the birth hospitalization as if this werea known case of hemolysis; (2) during the birth hospitalization initiate the evaluation as given in thegraphic or as suggested by hematology consultation; and (3) consider that the neonate could havemore severe jaundice than the parent did as a neonate, particularly if there has been coinheritanceof a polymorphism-retarding bilirubin uptake or conjugation. CBC, complete blood cell count.
1110 CHRISTENSEN et alby guest on August 17, 2016Downloaded from
which these methods were proveneffective with the blood of neonates.Sodium dodecyl sulfate polyacrylamidegel electrophoresis has been used aswell but has generally been replacedby other methods.31
Table 2 lists laboratory evaluationswe consider to be helpful indiagnosing HS in a neonate.Sequencing of the relevant genes canbe performed as a confirmatory test(available in a few referencelaboratories) when desired.32 Wereserve genetic sequencing for caseswith no family history and a severeHS phenotype. The goal would be tomake a clear diagnosis, allow forappropriate therapy, and to provideparents and family members withgenetic information for counselingregarding risk of recurrence.
TREATMENT
Phototherapy should reduce thebilirubin level of jaundiced neonates
with HS, and it is the mainstay oftreatment in the first days afterbirth.33–35 When a bilirubin level isfound to be in the high or high tointermediate risk zone (.75thpercentile reference interval),phototherapy should be providedimmediately. An exchange transfusionfollowed by intensive phototherapyshould follow American Academy ofPediatrics’ guidelines.20,21,36
When signs of anemia appear, packederythrocyte transfusions are helpful.Longitudinal studies indicate thattransfusion requirements abate inmost patients by 1 year of age. Thefew patients who remain transfusiondependent (typically those withsevere anemia in utero orimmediately after birth) experiencesevere HS. In a few reports,recombinant erythropoietin (rEPO, orlong-acting darbepoetin rEPO)therapy has been used as analternative or adjunct totransfusion.37–39 The rationale for
rEPO treatment relates to the relativehypoplastic phase of erythropoiesisduring the first weeks to months afterbirth. This phase might be associatedwith the abrupt decrease after birthfrom the highly stimulatederythropoiesis during fetal life, theswitch of erythropoietin (EPO)production from the liver to thekidney, the switch from fetal to adulthemoglobin, or a lower serum level ofEPO in infants compared with olderchildren. Patients with moderateor severe HS should receive folatesupplementation to preventcomplications of folic acid deficiency.Splenectomy is rarely undertakenin the first year of life. Because
FIGURE 5Evaluating a neonate with problematic jaundice in whom the etiology is unclear. Not all neonateswho receive phototherapy for $2 days have hemolytic jaundice. However, if hemolytic jaundice issuspected, the algorithm given here for stepwise evaluation of the etiology might be useful. CBC,complete blood cell count; G6PD, glucose-6-phosphate dehydrogenase deficiency.
FIGURE 6A, Results of Osmotic Fragility testing of a neo-nate with HS. The diagnosis is supported byfinding that red blood cells are more fragile toosmotic lysis than normal (the normal range isshown by the 2 dotted lines). HS cells undergomore hemolysis than normal as the sodiumchloride (NaCl) concentration is increased. B,Results of Eosin-5-Maleimide Fluorescent Cyto-metric (EMA-FITC) testing of the same neonatewith HS shown in panel (A). The diagnosis issupported by finding that a large fraction ofthe red blood cells have reduced EMA binding,with a leftward displacement of events, com-pared with the normal range (footprint). Sidescatter (SSC) reflects cell size.
PEDIATRICS Volume 135, number 6, June 2015 1111by guest on August 17, 2016Downloaded from

hemolysis abates in most patients,careful symptomatic management isprudent, with transfusion therapy asindicated.
NATURAL HISTORY OF HS DURINGINFANCY
This topic has received relatively littleattention in published series orreviews.40–45 Delhommeau et al10
reported on 34 infants with HSduring their first year of life, andneonatal jaundice was present invirtually all of the infants. Twenty-seven infants were treated withphototherapy, and 3 receivedexchange transfusions in the firstweek due to hyperbilirubinemia.Thirty-one of the 34 infants hadpallor and dyspnea during the firstmonth. Twenty-six (76%) required$1 red blood cell transfusion duringthe first year; 12 had a singletransfusion, and 14 had $2transfusions. The authors’ practicewas to order transfusions if thehemoglobin concentration fell to,10 g/dL in the first week and,8.5 g/dL thereafter. Transfusionswere rarely needed in the first week oflife but were commonly given in month2 of life. Only 8 (24%) of those withHS continued to receive transfusionsafter 6 months of age. Six underwentsplenectomies at 2 to 5 years of age.
The natural history of HS during thefirst week of life can involvehyperbilirubinemia, which is
sometimes severe, leading tobilirubin-induced neurologicdysfunction.6,7,13,43 In the USAKernicterus Registry, 23 of the 125patients with kernicterus hada hemolytic condition, of whom 3 hadHS and severe icteric sequelae.13
There should be close observation forhematologic decompensation duringacute illnesses. Infants with HS mayexperience hemolytic or aplasticcrises, similar to children and adultswith HS. This outcome is particularlytrue after 6 months of age whenmaternally derived immunoglobulinG antibodies to microbes have waned.Parvovirus B19–mediated aplasia isa common occurrence in childrenwith HS.45 Parents should beprovided anticipatory guidanceregarding the occurrence ofhematologic decompensationaccompanying systemic illness. Thewide spectrum of disease severityrequires individualized plans forfrequency of follow-up visits. Thesevisits should assess the degree ofanemia and monitor growth anddevelopment. Children with HS whocontinue to require bloodtransfusions should be closelymonitored for iron overload.
CONCLUSIONS
A working knowledge of thepresentation, diagnosis, andtreatment of HS is essential for thoseproviding care to neonates. When
a newborn infant has significanthyperbilirubinemia, a careful reviewof the family history, evaluation of thered blood cell indices, andinterpretation of the peripheral bloodsmear may be all that is needed tomake a diagnosis of HS. In selectedcases measuring erythrocyte EMAbinding, incubated osmotic fragilityor flow cytometric detection oferythrocyte osmotic fragility may behelpful. In problematic cases,hematologic consultation and DNAsequencing can provide the diagnosis.In any newborn infant with hemolyticjaundice, rigorous bilirubinmonitoring and treatment are needed,particularly during the first weeks oflife, and monitoring of thehemoglobin level is required duringthe first months of life. If health careproviders develop an early suspicionof HS, establish the diagnosis, provideappropriate treatment, and offeranticipatory guidance to homecaregivers, adverse outcomes can beavoided.
REFERENCES
1. Gallagher PG. Abnormalities of theerythrocyte membrane. Pediatr ClinNorth Am. 2013;60(6):1349–1362
2. Sangerman J, Maksimova Y, Edelman EJ,Morrow JS, Forget BG, Gallagher PG.Ankyrin-linked hereditary spherocytosisin an African-American kindred. Am JHematol. 2008;83(10):789–794
3. Sheffield MJ, Christensen RD. Evaluatingneonatal hyperbilirubinemia in late
TABLE 2 Laboratory Evaluation for HS in a Jaundiced Neonate
HS Ratio (MCHC/MCV) EMA Flowa Incubated Osmotic Fragility DNA Sequencingb
Neonates with HS willgenerally have a highMCHC and a low MCV,producing an elevatedratio (.0.36)
EMA dye binds stoichiometrically toband 3 and Rh-related membraneproteins. Decreased fluorescenceintensity of EMA-tagged erythrocytesdue to loss of membrane proteins isseen in HS. Decreased EMA binding isalso seen in other disorders such ashereditary pyropoikilocytosis andcongenital dyserythropoietic anemia II
HS erythrocytes are more susceptible toosmotic lysis than normal erythrocytesdue to decreased membrane surfacearea. Incubation overnight stressesthe already fragile HS erythrocyte,accentuating the defect. Spherocytesfrom any cause, including ABOincompatibility, will yield a positiveresult on osmotic fragility testing
Not needed to diagnosis most cases of HS.However, it can establish the diagnosis indifficult cases. Consider sequencing ofrelevant genes when family history isnegative and severe DAT-negative hemolysisis idiopathic
a Available in many reference laboratories.b Available in selected hematology/genetics reference laboratories. For example: Blood Disease Reference Hematology Laboratory, http://www.yaleblooddiseaselab.org/, 310 Cedar Street,CB 541a, New Haven, CT 06520, Tel. (203)737-1349, Fax (203)785-3896; ARUP Laboratories, http://www.aruplab.com/testing, 500 Chipeta Way, Salt Lake City, UT 84108, Tel. (800)522-2787, Fax(800)522-2706; GeneDx, http://www.genedx.com, 207 Perry Parkway Gaithersburg, MD 20877, Tel. (301)519-2100, Fax (301)519-2892; and Mayo Medical Laboratories, http://www.mayo-medicallaboratories.com/, 3050 Superior Drive NW, Rochester, MN 55901, Tel. (800)553-1710, Fax (507)284-1759.
1112 CHRISTENSEN et alby guest on August 17, 2016Downloaded from

preterm Hispanic twins led to thediagnosis of hereditary spherocytosis inthem, and in their sibling and in theirmother. J Perinatol. 2011;31(9):625–627
4. Crisp RL, Solari L, Vota D, et al. Aprospective study to assess thepredictive value for hereditaryspherocytosis using five laboratory tests(cryohemolysis test, eosin-59-maleimideflow cytometry, osmotic fragility test,autohemolysis test, and SDS-PAGE) on 50hereditary spherocytosis families inArgentina. Ann Hematol. 2011;90(6):625–634
5. Perrotta S, Gallagher PG, Mohandas N.Hereditary spherocytosis. Lancet. 2008;372(9647):1411–1426
6. Iolascon A, Faienza MF, Moretti A,Perrotta S, Miraglia del Giudice E. UGT1promoter polymorphism accounts forincreased neonatal appearance ofhereditary spherocytosis. Blood. 1998;91(3):1093
7. Berardi A, Lugli L, Ferrari F, et al.Kernicterus associated with hereditaryspherocytosis and UGT1A1 promoterpolymorphism. Biol Neonate. 2006;90(4):243–246
8. Qader A, Ismail AQ, Gandhi A, El-Shimy N.Intractable neonatal jaundice due tohereditary spherocytosis and Gilbert’ssyndrome. BMJ Case Rep. 2011 Jul 28;2011
9. Korkmaz U, Duman AE, O�gütmen Koç D,et al. Severe jaundice due to coexistenceof Dubin-Johnson syndrome andhereditary spherocytosis: a case report.Turk J Gastroenterol. 2011;22(4):422–425
10. Delhommeau F, Cynober T,Schischmanoff PO, et al. Natural historyof hereditary spherocytosis during thefirst year of life. Blood. 2000;95(2):393–397
11. Christensen RD, Henry E. Hereditaryspherocytosis in neonates withhyperbilirubinemia. Pediatrics. 2010;125(1):120–125
12. Chavez-Bueno S, Beasley JA, GoldbeckJM, et al. ‘Haptoglobin concentrationsin preterm and term newborns’.J Perinatol. 2011;31(7):500–503
13. Johnson L, Bhutani VK, Karp K, Sivieri EM,Shapiro SM. Clinical report from thepilot USA Kernicterus Registry (1992 to2004). J Perinatol. 2009;29(suppl 1):S25–S45
14. Christensen RD, Lambert DK, Henry E,et al. Unexplained extremehyperbilirubinemia among neonates ina multihospital healthcare system. BloodCells Mol Dis. 2013;50(2):105–109
15. Sgro M, Campbell D, Shah V. Incidenceand causes of severe neonatalhyperbilirubinemia in Canada. CMAJ.2006;175(6):587–590
16. Saada V, Cynober T, Brossard Y, et al.Incidence of hereditary spherocytosis ina population of jaundiced neonates.Pediatr Hematol Oncol. 2006;23(5):387–397
17. Christensen RD, Yaish HM, Lemons RS.Neonatal hemolytic jaundice:morphologic features of erythrocytesthat will help you diagnose theunderlying condition. Neonatology. 2014;105(4):243–249
18. Yaish HM, Christensen RD, Henry E, BaerVL, Bennett ST. A simple method ofscreening newborn infants forhereditary spherocytosis. J AppliedHematol. 2013:27–32
19. Mullier F, Lainey E, Fenneteau O, et al.Additional erythrocytic and reticulocyticparameters helpful for diagnosis ofhereditary spherocytosis: results ofa multicentre study. Ann Hematol. 2011;90(7):759–768
20. American Academy of PediatricsSubcommittee on Hyperbilirubinemia.Management of hyperbilirubinemia inthe newborn infant 35 or more weeks ofgestation. Pediatrics. 2004;114(1):297–316
21. Maisels MJ, Bhutani VK, Bogen D,Newman TB, Stark AR, Watchko JF.Hyperbilirubinemia in the newborninfant . or =35 weeks’ gestation: anupdate with clarifications. Pediatrics.2009;124(4):1193–1198
22. Jarolim P, Murray JL, Rubin HL, et al.Characterization of 13 novel band 3 genedefects in hereditary spherocytosis withband 3 deficiency. Blood. 1996;88(11):4366–4374
23. King MJ, Zanella A. Hereditary red cellmembrane disorders and laboratorydiagnostic testing. Int J Lab Hematol.2013;35(3):237–243
24. Christensen RD, Agarwal AM,Nussenzveig RH, Heikal N, Liew MA, YaishHM. Evaluating eosin-5-maleimidebinding as a diagnostic test for
hereditary spherocytosis in newborninfants [published online ahead of printNovember 6, 2014]. J Perinatol. doi: doi:10.1038/jp.2014.202
25. Bolton-Maggs PH, Langer JC, Iolascon A,Tittensor P, King MJ; GeneralHaematology Task Force of the BritishCommittee for Standards inHaematology. Guidelines for thediagnosis and management ofhereditary spherocytosis—2011 update.Br J Haematol. 2012;156(1):37–49
26. Schröter W, Kahsnitz E. Diagnosis ofhereditary spherocytosis in newborninfants. J Pediatr. 1983;103(3):460–463
27. Trucco JI, Brown AK. Neonatalmanifestations of hereditaryspherocytosis. Am J Dis Child. 1967;113(2):263–270
28. Won DI, Suh JS. Flow cytometricdetection of erythrocyte osmotic fragility.Cytometry B Clin Cytom. 2009;76(2):135–141
29. Warang P, Gupta M, Kedar P, Ghosh K,Colah R. Flow cytometric osmoticfragility—an effective screeningapproach for red cell membranopathies.Cytometry B Clin Cytom. 2011;80(3):186–190
30. Crisp RL, Solari L, Gammella D,Schvartzman GA, Rapetti MC, Donato H.Use of capillary blood to diagnosehereditary spherocytosis. Pediatr BloodCancer. 2012;59(7):1299–1301
31. Bianchi P, Fermo E, Vercellati C, et al.Diagnostic power of laboratory tests forhereditary spherocytosis: a comparisonstudy in 150 patients grouped accordingto molecular and clinical characteristics.Haematologica. 2012;97(4):516–523
32. Christensen RD, Nussenzveig RH, YaishHM, Henry E, Eggert LD, Agarwal AM.Causes of hemolysis in neonates withextreme hyperbilirubinemia. J Perinatol.2014;34(8):616–619
33. Maisels MJ, McDonagh AF. Phototherapyfor neonatal jaundice. N Engl J Med.2008;358(9):920–928
34. Johnson L, Bhutani VK. The clinicalsyndrome of bilirubin-inducedneurologic dysfunction. Semin Perinatol.2011;35(3):101–113
35. Dennery PA, Seidman DS, Stevenson DK.Neonatal hyperbilirubinemia. N Engl JMed. 2001;344(8):581–590
PEDIATRICS Volume 135, number 6, June 2015 1113by guest on August 17, 2016Downloaded from

36. Flaherman VJ, Kuzniewicz MW, EscobarGJ, Newman TB. Total serum bilirubinexceeding exchange transfusionthresholds in the setting of universalscreening. J Pediatr. 2012;160(5):796–800.e1
37. Tchernia G, Delhommeau F, Perrotta S,et al; ESPHI working group on hemolyticanemias. Recombinant erythropoietintherapy as an alternative to bloodtransfusions in infants with hereditaryspherocytosis. Hematol J. 2000;1(3):146–152
38. Neuman-Łaniec M, Wierzba J, Irga N,Wasilewska E, Balcerska A. Recombinanterythropoietin—an alternative therapyto red cell blood transfusions in infantswith hereditary spherocytosis [inPolish]. Przegl Lek. 2002;59(10):871–872
39. Schiff M, Haÿs S, Sann L, Putet G.Recombinant human erythropoietin (r-HuEPO) therapy in a newborn withhereditary spherocytosis [in French].Arch Pediatr. 2003;10(4):333–336
40. Delaunay J, Nouyrigat V, Proust A, et al.Different impacts of alleles alphaLEPRA
and alphaLELY as assessed versus a novel,virtually null allele of the SPTA1 gene intrans. Br J Haematol. 2004;127(1):118–122
41. Shapiro CM, Josephson AM, RozengvaigS, Kauffman A. Hereditary spherocytosisin the neonatal period: diagnosis,incidence, and treatment. J Pediatr. 1957;50(3):308–314
42. Christensen RD, Yaish HM, NussenzveigRH, et al. Acute kernicterus in a neonate
with O/B blood group incompatibility anda mutation in SLC4A1. Pediatrics. 2013;132(2). Available at: www.pediatrics.org/cgi/content/full/132/2/e531
43. Bogardus HH, Maksimova YD, Forget BG,Gallagher PG. A de novo band 3 mutationin hereditary spherocytosis. PediatrBlood Cancer. 2012;58(6):1004
44. Gundel F, Eber S, Heep A. A new ankyrinmutation (ANK1 EXON E9X) causingsevere hereditary spherocytosis in theneonatal period. Ann Hematol. 2011;90(2):231–232
45. Forde DG, Cope A, Stone B. Acuteparvovirus B19 infection in identicaltwins unmasking previously unidentifiedhereditary spherocytosis. BMJ Case Rep.2014 Jul 29;2014
GETTINGTHELASTDROP:Ourrefrigerator is litteredwithpartiallyfilledbottlesofmustard, ketchup, and mayonnaise. In order to get the last of the ingredient, I willstore them in such a way that promotes flow of the material toward the nozzle.Despitemy best efforts, however, far toomuchmustard or ketchup remains stuck inthe bottle and unusable. It would appear that others have the same problem.Consumer Reports magazine reported in September 2009 that a considerableamount of what we buy, from toothpaste to laundry detergent, cannot be used as itremains trapped in the container. Their tests showed that 25 percent of skin lotionand 15 percent of condiments such asmustard and ketchup are thrown away. Help,however, may be on the way.As reported in The New York Times (Science: March 23, 2015), a MassachusettsInstitute of Technology professor and one of his graduate students have developeda coating for the inside of containers to make them slippery. When a fluid movesthrough a pipe or a container, the layer next to the pipe usually sticks to it and doesnotmove. Thefluid farthest from thewall of the pipe or containermoves fastest. Thetrick is tomake the boundarybetween thewall of the container and thefirst layer offluid slippery with a lubricant. The MIT scientists developed an equation to predictthe interactions between the surface of the container, the lubricant, and air andwater. The lubricant bindsmore strongly to the surface of the container than to theliquid, allowing the liquid to slideona layerof lubricant rather thanover the surfaceof the container. The first successful application of the equation was in developinga lubricant that allows glue to flow effortlessly through a glue bottle. Thematerialscan be modified for different applications. For instance, specific coatings can bedeveloped to help crude oil, toothpaste, and honey flow more easily in theircontainers.While the industrial applications are impressive, I am particularly happy that I willnot be wasting as much food in the future; I expect food applications to appear in2017.
Noted by WVR, MD
1114 CHRISTENSEN et alby guest on August 17, 2016Downloaded from

DOI: 10.1542/peds.2014-3516; originally published online May 25, 2015; 2015;135;1107Pediatrics
Robert D. Christensen, Hassan M. Yaish and Patrick G. GallagherSpherocytosis in Neonates
A Pediatrician's Practical Guide to Diagnosing and Treating Hereditary
ServicesUpdated Information &
/content/135/6/1107.full.htmlincluding high resolution figures, can be found at:
References
/content/135/6/1107.full.html#ref-list-1at:This article cites 41 articles, 9 of which can be accessed free
Rs)3Peer Reviews (PPost-Publication
/cgi/eletters/135/6/1107
R has been posted to this article: 3One P
Subspecialty Collections
/cgi/collection/blood_disorders_subBlood Disorders
/cgi/collection/hematology:oncology_subHematology/Oncologythe following collection(s):This article, along with others on similar topics, appears in
Permissions & Licensing
/site/misc/Permissions.xhtmltables) or in its entirety can be found online at: Information about reproducing this article in parts (figures,
Reprints /site/misc/reprints.xhtml
Information about ordering reprints can be found online:
rights reserved. Print ISSN: 0031-4005. Online ISSN: 1098-4275.Grove Village, Illinois, 60007. Copyright © 2015 by the American Academy of Pediatrics. All and trademarked by the American Academy of Pediatrics, 141 Northwest Point Boulevard, Elkpublication, it has been published continuously since 1948. PEDIATRICS is owned, published, PEDIATRICS is the official journal of the American Academy of Pediatrics. A monthly
by guest on August 17, 2016Downloaded from

DOI: 10.1542/peds.2014-3516; originally published online May 25, 2015; 2015;135;1107Pediatrics
Robert D. Christensen, Hassan M. Yaish and Patrick G. GallagherSpherocytosis in Neonates
A Pediatrician's Practical Guide to Diagnosing and Treating Hereditary
/content/135/6/1107.full.html
located on the World Wide Web at: The online version of this article, along with updated information and services, is
of Pediatrics. All rights reserved. Print ISSN: 0031-4005. Online ISSN: 1098-4275.Boulevard, Elk Grove Village, Illinois, 60007. Copyright © 2015 by the American Academy published, and trademarked by the American Academy of Pediatrics, 141 Northwest Pointpublication, it has been published continuously since 1948. PEDIATRICS is owned, PEDIATRICS is the official journal of the American Academy of Pediatrics. A monthly
by guest on August 17, 2016Downloaded from