a method for identifying the viral genes required for herpesvirus dna replication
TRANSCRIPT

A Method for Identifying the Viral Genes Required for Herpesvirus DNA ReplicationAuthor(s): Mark D. ChallbergSource: Proceedings of the National Academy of Sciences of the United States of America,Vol. 83, No. 23 (Dec. 1, 1986), pp. 9094-9098Published by: National Academy of SciencesStable URL: http://www.jstor.org/stable/28895 .
Accessed: 07/05/2014 19:09
Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at .http://www.jstor.org/page/info/about/policies/terms.jsp
.JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range ofcontent in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new formsof scholarship. For more information about JSTOR, please contact [email protected].
.
National Academy of Sciences is collaborating with JSTOR to digitize, preserve and extend access toProceedings of the National Academy of Sciences of the United States of America.
http://www.jstor.org
This content downloaded from 169.229.32.136 on Wed, 7 May 2014 19:09:24 PMAll use subject to JSTOR Terms and Conditions

Proc. Natl. Acad. Sci. USA Vol. 83, pp. 9094-9098, December 1986 Genetics
A method for identifying the viral genes required for herpesvirus DNA replication
(plasmid transfection/transient assay/herpes simplex virus oriL and oris)
MARK D. CHALLBERG
Laboratory of Viral Diseases, National Institute of Allergy and Infectious Diseases, 9000 Rockville Pike, Bethesda, MD 20894
Communicated by Daniel Nathans, August 13, 1986
ABSTRACT Several laboratories have shown that transfected plasmid DNAs containing either of the two known origins of herpes simplex virus (HSV) DNA replication, oris or oriL, are replicated in HSV-1-infected cells or in cells cotrans- fected with virion DNA. I have found that HSV-1 (KOS) DNA digested to completion with the restriction enzyme Xba I is as efficient as intact viral DNA in supporting the in vivo replica- tion of cotransfected plasmids containing oris. On the basis of this result, several of the Xba I restriction fragments of HSV-1 DNA were cloned into the plasmid vector pUC19, and combi- nations of cloned DNAs were tested for their ability to supply the trans-acting functions required for HSV origin-dependent replication. A combination of fi"ve cloned fragments of HSV-1 can supply all of the necessary functions: Xba I C (coordinates 0.074-0.294), Xba I F (coordinates 0.294-0.453), Xba I E (coordinates 0.453-0.641), Xba I D (coordinates 0.641-0.830), and EcoRI JK (coordinates 0.0-0.086; 0.830-0.865). Tran- sient plasmid replication in this system is dependent on the presence of either oris or oriL in cis. The plasmid containing Xba I F can be replaced by two smaller plasmids, one of which contains only the gene for the HSV-encoded DNA polymerase, and the other of which contains only the gene for the major DNA binding protein (ICP8). Thus, plasmid DNA replication in this system depends on two of the genes known from genetic studies to be essential for viral DNA replication in infected cells. This system defines a simple complementation assay for cloned fragments of HSV DNA that contain other genes involved in viral DNA replication and should lead to the rapid identifica- tion of all such genes.
Herpes simplex virus (HSV) is a potentially useful model for studying DNA replication in eukaryotic cells. The viral genome is a linear duplex DNA molecule of about 150 kilobases (kb) (1). Replication takes place within the nucleus of the infected cell, and the available genetic and biochemical evidence suggests that many, if not all, of the proteins involved in viral DNA replication are virus-encoded. Thus, in principle, HSV DNA replication should be amenable to the combined biochemical-genetical approach that has proven so powerful in the dissection of the process of DNA replication in prokaryotic systems.
Although the overall mechanism of HSV DNA replication is not yet well-understood, several important features have emerged. Three cis-acting elements that are thought to function as origins of replication have been identified (2-6). The existence of these origins was inferred from the structure of defective viral genomes (2, 3), and several laboratories have shown that plasmids containing these sequences are replicated when they are introduced into HSV-infected cells (2-6). Two of these origins, both named on's, are identical, since they are located within the innverted repeat sequence
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. ?1734 solely to indicate this fact.
flanking the short component of the genome (2-5). The other origin, named oriL, is located within the unique sequences of the long component (2, 3, -6). Following initiation, DNA synthesis proceeds by way of intermediates in which the genomic termini are joined (7-9). The structure of these intermediates has not been precisely defined; they may consist of circular or concatemeric molecules, or both (7-9). Unit length linear genomes are subsequently formed during the packagirng of DNA into virions.
There are several viral gene products that have been shown by genetic means to be essential for DNA replication in cultured cells. These include a DNA polymerase (Pol; refs. 10-14), a single-stranded DNA binding protein (DBP; refs. 15-18), a ribonucleotide reductase (RR; ref. 19), and an exonuclease (Exo; ref. 20). These four proteins, which are thought to be directly involved in DNA replication, are the products of early genes. Another viral gene product, the immediate early protein IE175, is also required for DNA replication (21, 22); however, since the transcription of all early genes depends on the prior expression of one or more immediate early genes (23, 24), including IE175, the role of IE175 in]DNA replication may be indirect. In addition to these known proteins, it is likely that other viral gene products are also involved in viral DNA replication since other complementation groups of temperature-sensitive mu- tants that are unable to carry out DNA replication at the nonpermissive temperature have been reported (25, 26).
In this paper, I describe the development of a system for identifying segments of the HSV genome that encode pro- teins necessary for viral DNA replication. I show that a combination of several cloned restriction fragments of HSV DNA, collectively containing the majority of the viral DNA sequence, will supply all of the functions required for the replication of plasmids containing HSV replication origins when cotransfected into Vero cells. This approach defines a simple complementation assay for trans-acting HSV genes required for viral DNA replication as well as any additional genes necessary for their expression. In view of the relatively simple organization of HSV genes (27, 28), this assay should now make it possible to systematically identify all of the viral genes involved in HSV DNA replication.
MATERIALS AND METHODS Cells and Virus. Vero cells were propagated in Eagle's
minimal essential medium containing 10% fetal calf serum. The KOS strain of HSV-1 was used in this study and virus stocks were grown and assayed in Vero cells.
DNA Isolation. HSV-1 DNA was isolated by the procedure of Denniston et al. (29) from Vero cells infected at a multiplicity of infection of 0.001 plaque-forming unit per cell. Bacterial plasmid DNAs were isolated by the method of
Abbreviations: HSV, herpes simplex virus; Pol, DNA polymerase; DBP, DNA binding protein; RR, ribonucleotide reductase; Exo, exonuclease; kb, kilobase(s); bp, base pair(s).
9094
This content downloaded from 169.229.32.136 on Wed, 7 May 2014 19:09:24 PMAll use subject to JSTOR Terms and Conditions

Genetics: Challberg Proc. Natl. Acad. Sci. USA 83 (1986) 9095
Birnboim and Doly (30) and were further purified by banding in cesium chloride/ethidium bromide gradients.
Construction of Recombinant Plasmids. All recombinant plasmids containing HSV DNA were derived from the KOS strain of HSV-1, and plasmids were introduced into Esche- richia coli strain DH5 by the procedure of Hanahan (31).
pMC110. pKOS-BX (obtained from S. Weller), containing the BamHI X fragment of HSV-1 inserted into the BamHI site of pBR325, was digested with Sma I and a 230-base-pair (bp) fragment containing oris was purified by agarose gel electro- phoresis. This fragment was' inserted into the Sma I site of pUC19 (32). The structure of the recombinant clone was verified by sequencing the 230-bp insertion.
pMC121, -122, -123, and -124. HSV-1 DNA was incubated with the Klenow fragment of Pol I in the presence of the four dNTPs to form blunt ends at the genomic termini. This DNA' was then ligated to a 10-fold molar excess of Xba I linker oligonucleotide (New England Biolabs) and digested with Xba I. The digested DNA was fractioned by agarose gel electrophoresis using low-melting-temperature agarose (Bethesda Research Laboratories). DNA fragments =20-30 kb in size were excised from the gel and purified by extraction of the melted gel slice with phenol and chloroform, followed by ethanol precipitation. This DNA was then joined, using T4 DNA ligase, to Xba I-cleaved pUC19 DNA that had been treated with calf intestine phosphatase, and the ligation mixture was used to transform E. coli strain DH5. Trans- formed colonies were screened by hybridization with one of four nick-translated fragments of HSV DNA: EcoRI D, EcoRI F, EcoRI A, and EcoRI EK. Plasmid DNA was isolated from, several colonies which hybridized to the individual probes, and plasmid structure was analyzed by digestion with several different restriction enzymes, includ- ing BamHI, EcoRI, Hindlll, and Hpa I. Approximately 50% of the hybridizing colonies had plasmid' DNA with the predicted restriction enzyme patterns; the remaining 50% had large deletions or other rearrangements. Purified prepara- tions of pMC122 (containing Xba I F) are heterogeneous; -50% of the pMC122 DNA in the preparation used in this work contained small deletions in the oriL region.
pNNI and pNN3. Subclones of the Xba I F region were constructed starting with plasmid pMC119, which contains the Hpa I B fragment of HSV-I DNA cloned into the Sma I site of pUC19. Plasmid DNA from single colonies of DH5 containing pMC119 was digested with BamHI to determine the structure of the DNA in the intragenic region between dbp and pol (6). Several colonies contained a single species of plasmid DNA with a 150-bp deletion in that region, and DNA from one of these clones was used as the starting material in the construction of pNN1 and pNN3. Detailed restriction mapping suggested that the end points of the 150-bp deletion are identical to those of a similarly sized deletion described by Weller et al. (6). This is the most common stable deletion in the oriL region of plasmids derived from the KOS strain of HSV-1 (S. Weller, personal communication). pNN1 was constructed by isolating the 6.5-kb Sac I fragment containing the dbp gene from pMC119 and ligating this fragment to Sac I-digested pUC19. pNN3 was constructed as follows: pMC119 was digested with Pvu I and Xba I. The DNA was incubated with the Klenow fragment of PolI and dNTPs to form blunt ends, and a 9-kb fragment containing the pol gene was isolated by gel electrophoresis. This fragment was ligated into the Sma I site of pUC19 to form pNN2. pNN2 was partially digested with Kpn I and completely digested with HindIII, and a 5.5-kb fragment containing the pol gene was cloned between the Kpn I and HindIII sites of pUC19. The locations of restriction sites within the genes for Pol and DBP were deduced from the published DNA sequences (33, 34).
Assay for Plasmid DNA Replication. Vero cells were plated in 28-cm2 dishes at a density of about 3 x 106 cells per dish
and incubated for 14-16 hr at 37?C, at which time they were nearly confluent. Cells were transfected using the calcium phosphate coprecipitation technique (35, 36) as follows. Three to 4 hr before transfection, the medium was replaced. Combinations of HSV DNA and/or plasmid DNAs (0.5 Ag each) were mixed with sonicated calf thymus DNA and 2.0 M CaCl2 so that the final concentrations were 20 ,ug/ml of DNA and 0.25 M CaCl2 in a volume of 0.25 ml. This mixture was added dropwise to 0.25 ml of 2x concentrated HBS (HBS = 25 mM Hepes/140 mM NaCl/1.4 mM Na2PO4) and incubated at room temperature for 20-30 min, at which point it was added directly to the medium of the cells. After 4 hr at 37?C, the medium was removed and 0.5 ml of 15% glycerol in HBS was added. After 3-4 min, the cells were washed with isotonic buffer and the medium was replaced. After 14-16 hr at 37?C the medium was removed and the cells were lysed by the addition of 2.0 ml of 10 mM Tris HCl, pH 8.0/10 mM EDTA/2% NaDodSO4/100 ,Ag of proteinase K per ml. The cell lysate was incubated at 37?C for 2-8 hr, after which sodium acetate was added to a concentration of 0.3 M, and the resulting solution was extracted once each with phenol and chloroform/isoamyl alcohol (24:1).- RNase A was added at a concentration of 50 Ag/ml. Following incubation at 37?C for 30 min, DNA was precipitated with ethanol and redis- solved in 10 mM Tris-HCl, pH 8.0/1 mM EDTA (TE) containing 50 Ag of RNase A per ml. One-fourth of the DNA from a dish was digested with EcoRI, Dpn I, and, in some experiments, HindIII, in a reaction mixture (0.1 ml) contain- ing 10 mM Tris HCl (pH 7.5), 150 mM NaCl, 10 mM MgCl2, 6 mM 2-mercaptoethanol, and 10 units of each restriction enzyme for 4-14 hr at 37?C. The DNA was precipitated with ethanol, dissolved in TE, and fractionated by gel electropho- resis in a 1% agarose gel. The DNA in the gel was transferred to nitrocellulose and hybridized with 32P-labeled pUC19 DNA (1-3 x 108 dpm/,g) as described by Maniatis et al. (37). DNA fragments on the filter that hybridized to the probe were located by autoradiography for 5-24 hr using Kodak XAR film and an intensifying screen.
RESULTS
Xba I Does Not Cleave Any HSV-1 Gene That Is Essential for DNA Replication. As mentioned above, it has been shown previously that cells transfected with purified HSV virion DNA will support the replication of cotransfected plasmids that contain oris or oriL (2-6). It seems likely that plasmid replication in such a system depends on the expression of only a limited subset of viral genes-namely, those genes directly involved in replication of viral DNA and any regu- latory genes necessary for their expression. As a first step in locating essential replication genes, HSV-1 DNA was digest- ed with various restriction enzymes to test whether the resulting fragments could supply the trans-acting functions required for HSV origin-specific replication of transfected plasmid DNA.
Vero cell monolayers were transfected with a mixture of the plasmid pMC110, containing oris, and HSV-1 DNA that had been digested to completion with one of a number of different restriction enzymes. At 14-16 hr following trans- fection, total cellular DNA was isolated and digested with EcoRI, which cleaves pMC110 once, and Dpn I. Digestion with Dpn I serves as the basis for an assay of plasmid DNA replication (38), since this enzyme cleaves only DNA that has been methylated by the dam methylation system of E. coli. Thus, input pMC110 is cleaved into several fragments by Dpn I, but replicated plasmid DNA is not cleaved. The digested DNA was fractionated by agarose gel electrophoresis, trans- ferred to nitrocellulose, and probed with 32P-labeled pUC19 DNA for the presence of Dpn I-resistant pMC11O.
This content downloaded from 169.229.32.136 on Wed, 7 May 2014 19:09:24 PMAll use subject to JSTOR Terms and Conditions
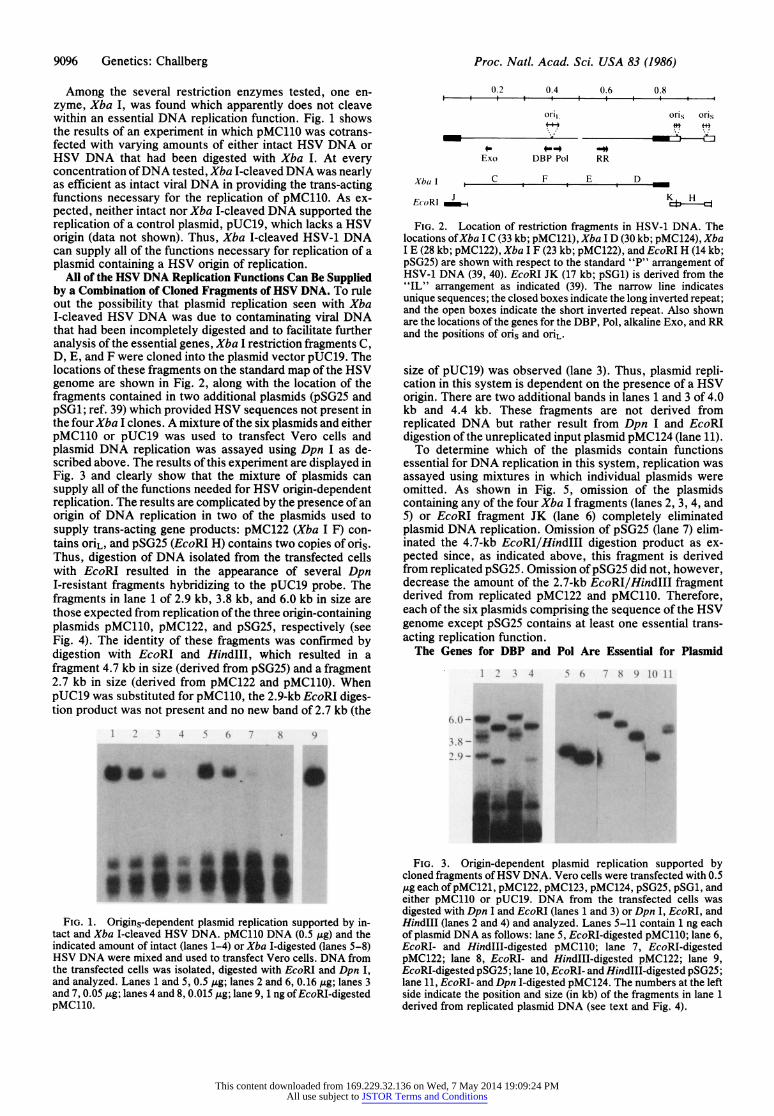
9096 Genetics: Challberg Proc. Natl. Acad. Sci. USA 83 (1986)
Among the several restriction enzymes tested, one en- zyme, Xba I, was found which apparently does not cleave within an essential DNA replication function. Fig. 1 shows the results of an experiment in which pMC110 was cotrans- fected with varying amounts of either intact HSV DNA or HSV DNA that had been digested with Xba I. At every concentration of DNA tested, Xba I-cleaved DNA was nearly as efficient as intact viral DNA in providing the trans-acting functions necessary for the replication of pMC110. As ex- pected, neither intact nor Xba I-cleaved DNA supported the replication of a control plasmid, pUC19, which lacks a HSV origin (data not shown). Thus, Xba I-cleaved HSV-1 DNA can supply all of the functions necessary for replication of a plasmid containing a HSV origin of replication.
All of the HSV DNA Replication Functions Can Be Supplied by a Combination of Cloned Fragments of HSV DNA. To rule out the possibility that plasmid replication seen with Xba I-cleaved HSV DNA was due to contaminating viral DNA that had been incompletely digested and to facilitate further analysis of the essential genes, Xba I restriction fragments C, D, E, and F were cloned into the plasmid vector pUC19. The locations of these fragments on the standard map of the HSV genome are shown in Fig. 2, along with the location of the fragments contained in two additional plasmids (pSG25 and pSG1; ref. 39) which provided HSV sequences not present in the four Xba I clones. A mixture of the six plasmids and either pMC11O or pUC19 was used to transfect Vero cells and plasmid DNA replication was assayed using Dpn I as de- scribed above. The results of this experiment are displayed in Fig. 3 and clearly show that the mixture of plasmids can supply all of the functions needed for HSV origin-dependent replication. The results are complicated by the presence of an origin of DNA replication in two of the plasmids used to supply trans-acting gene products: pMC122 (Xba I F) con- tains oriL, and pSG25 (EcoRI H) contains two copies of oris. Thus, digestion of DNA isolated from the transfected cells with EcoRI resulted in the appearance of several Dpn I-resistant fragments hybridizing to the pUC19 probe. The fragments in lane 1 of 2.9 kb, 3.8 kb, and 6.0 kb in size are those expected from replication of the three origin-containing plasmids pMC110, pMC122, and pSG25, respectively (see Fig. 4). The identity of these fragments was confirmed by digestion with EcoRI and HindIII, which resulted in a fragment 4.7 kb in size (derived from pSG25) and a fragment 2.7 kb in size (derived from pMC122 and pMC110). When pUC19 was substituted for pMC110, the 2.9-kb EcoRI diges- tion product was not present and no new band of 2.7 kb (the
1 2 3 4 5 6 7 8 9
FIG. 1. Origins-dependent plasmid replication supported by in- tact and Xba I-cleaved HSV DNA. pMC11O DNA (0.5 ,g) and the indicated amount of intact (lanes 1-4) or Xba I-digested (lanes 5-8) HSV DNA were mixed and used to transfect Vero cells. DNA from the transfected cells was isolated, digested with EcoRI and Dpn I, and analyzed. Lanes 1 and 5, 0.5 ,ug; lanes 2 and 6, 0.16 ,utg; lanes 3 and 7, 0.05 ,ug; lanes 4 and 8, 0.015 ,ug; lane 9, 1 ng of EcoRI-digested pMC110.
0.2 0.4 0.6 0.8 e I I I I , I ,
oril oris oris
Exo DBP Pol RR
Xba I C F E D
EcoRi l
FIG. 2. Location of restriction fragments in HSV-1 DNA. The locations of Xba I C (33 kb; pMC121), Xba I D (30 kb; pMC124), Xba I E (28 kb; pMC122), Xba I F (23 kb; pMC122), and EcoRI H (14 kb; pSG25) are shown with respect to the standard "P" arrangement of HSV-1 DNA (39, 40). EcoRI JK (17 kb; pSG1) is derived from the "IL" arrangement as indicated (39). The narrow line indicates unique sequences; the closed boxes indicate the long inverted repeat; and the open boxes indicate the short inverted repeat. Also shown are the locations of the genes for the DBP, Pol, alkaline Exo, and RR and the positions of oris and oriL.
size of pUC19) was observed (lane 3). Thus, plasmid repli- cation in this system is dependent on the presence of a HSV origin. There are two additional bands in lanes 1 and 3 of 4.0 kb and 4.4 kb. These fragments are not derived from replicated DNA but rather result from Dpn I and EcoRI digestion of the unreplicated input plasmid pMC124 (lane 11).
To determine which of the plasmids contain functions essential for DNA replication in this system, replication was assayed using mixtures in which individual plasmids were omitted. As shown in Fig. 5, omission of the plasmids containing any of the four Xba I fragments (lanes 2, 3, 4, and 5) or EcoRI fragment JK (lane 6) completely eliminated plasmid DNA replication. Omission of pSG25 (lane 7) elim- inated the 4.7-kb EcoRI/HindIII digestion product as ex- pected since, as indicated above, this fragment is derived from replicated pSG25. Omission of pSG25 did not, however, decrease the amount of the 2.7-kb EcoRI/HindIII fragment derived from replicated pMC122 and pMC110. Therefore, each of the six plasmids comprising the sequence of the HSV genome except pSG25 contains at least one essential trans- acting replication function.
The Genes for DBP and Pol Are Essential for Plasmid
1 2 3 4 5 6 7 8 9 10 11
6.0-
FIG. 3. Origin-dependent plasmid replication supported by cloned fragments of HSV DNA. Vero cells were transfected with 0.5 ,ug each of pMC121, pMC122, pMC123, pMC124, pSG25, pSG1, and either pMC110 or pUC19. DNA from the transfected cells was digested with Dpn I and EcoRI (lanes 1 and 3) or Dpn I, EcoRI, and HindIII (lanes 2 and 4) and analyzed. Lanes 5-11 contain 1 ng each of plasmid DNA as follows: lane 5, EcoRI-digested pMC110; lane 6, EcoRI- and HindIII-digested pMC110; lane 7, EcoRI-digested pMC122; lane 8, EcoRI- and HindIII-digested pMC122; lane 9, EcoRI-digested pSG25; lane 10, EcoRI- and HindIII-digested pSG25; lane 11, EcoRI- and Dpn I-digested pMC124. The numbers at the left side indicate the position and size (in kb) of the fragments in lane 1 derived from replicated plasmid DNA (see text and Fig. 4).
This content downloaded from 169.229.32.136 on Wed, 7 May 2014 19:09:24 PMAll use subject to JSTOR Terms and Conditions
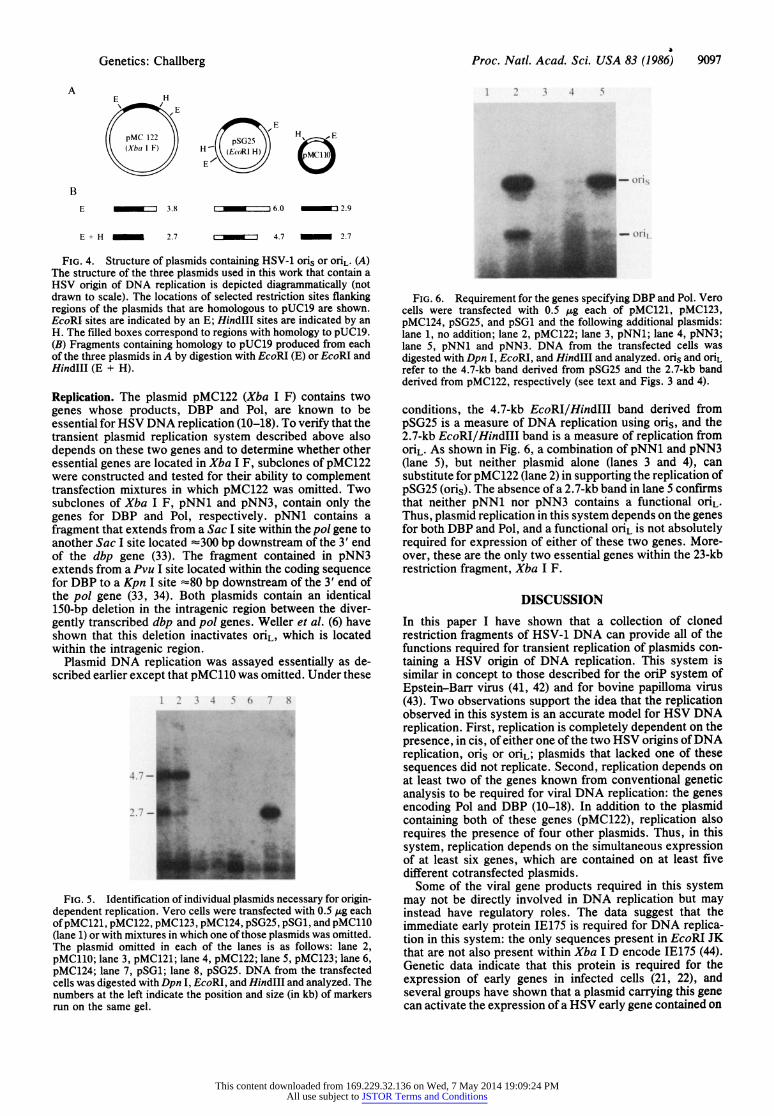
Genetics: Challberg Proc. Natl. Acad. Sci. USA 83 (1986) 9097
A E H
E
pMCl122j ((S2y H% ~E
B E 3.8 , 6.0 _ E 2.9
E + H 2.7 4.7 _ 2.7
FIG. 4. Structure of plasmids containing HSV-1 oris or oriL. (A) The structure of the three plasmids used in this work that contain a HSV origin of DNA replication is depicted diagrammatically (not drawn to scale). The locations of selected restriction sites flanking regions of the plasmids that are homologous to pUC19 are shown. EcoRI sites are indicated by an E; HindIII sites are indicated by an H. The filled boxes correspond to regions with homology to pUC19. (B) Fragments containing homology to pUC19 produced from each of the three plasmids in A by digestion with EcoRI (E) or EcoRI and HindIII (E + H).
Replication. The plasmid pMC122 (Xba I F) contains two genes whose products, DBP and Pol, are known to be essential for HSV DNA replication (10-18). To verify that the transient plasmid replication system described above also depends on these two genes and to determine whether other essential genes are located in Xba I F, subclones of pMC122 were constructed and tested for their ability to complement transfection mixtures in which pMC122 was omitted. Two subclones of Xba I F, pNN1 and pNN3, contain only the genes for DBP and Pol, respectively. pNN1 contains a fragment that extends from a Sac I site within the pol gene to another Sac I site located -300 bp downstream of the 3' end of the dbp gene (33). The fragment contained in pNN3 extends from a Pvu I site located within the coding sequence for DBP to a Kpn I site -80 bp downstream of the 3' end of the pol gene (33, 34). Both plasmids contain an identical 150-bp deletion in the intragenic region between the diver- gently transcribed dbp and pol genes. Weller et al. (6) have shown that this deletion inactivates OriL, which is located within the intragenic region.
Plasmid DNA replication was assayed essentially as de- scribed earlier except that pMC110 was omitted. Under these
1 2 3 4 5 6 7 8
4.7 -
2.7-_
FIG. 5. Identification of individual plasmids necessary for origin- dependent replication. Vero cells were transfected with 0.5 Ag each of pMC121, pMC122, pMC123, pMC124, pSG25, pSG1, and pMC110 (lane 1) or with mixtures in which one of those plasmids was omitted. The plasmid omitted in each of the lanes is as follows: lane 2, pMC110; lane 3, pMC121; lane 4, pMC122; lane 5, pMC123; lane 6, pMC124; lane 7, pSG1; lane 8, pSG25. DNA from the transfected cells was digested with Dpn I, EcoRI, and HindIII and analyzed. The numbers at the left indicate the position and size (in kb) of markers run on the same gel.
1 2 3 4 5
oris
I U-~~~~~~~~oriL
FIG. 6. Requirement for the genes specifying DBP and Pol. Vero cells were transfected with 0.5 ,ug each of pMC121, pMC123, pMC124, pSG25, and pSG1 and the following additional plasmids: lane 1, no addition; lane 2, pMC122; lane 3, pNN1; lane 4, pNN3; lane 5, pNN1 and pNN3. DNA from the transfected cells was digested with Dpn I, EcoRI, and HindIII and analyzed. oris and oriL
refer to the 4.7-kb band derived from pSG25 and the 2.7-kb band derived from pMC122, respectively (see text and Figs. 3 and 4).
conditions, the 4.7-kb EcoRI/HindIII band derived from pSG25 is a measure of DNA replication using oris, and the 2.7-kb EcoRI/HindIII band is a measure of replication from oriL. As shown in Fig. 6, a combination of pNN1 and pNN3 (lane 5), but neither plasmid alone (lanes 3 and 4), can substitute for pMC122 (lane 2) in supporting the replication of pSG25 (oris). The absence of a 2.7-kb band in lane 5 confirms that neither pNN1 nor pNN3 contains a functional oriL. Thus, plasmid replication in this system depends on the genes for both DBP and Pol, and a functional oriL is not absolutely required for expression of either of these two genes. More- over, these are the only two essential genes within the 23-kb restriction fragment, Xba I F.
DISCUSSION
In this paper I have shown that a collection of cloned restriction fragments of HSV-1 DNA can provide all of the functions required for transient replication of plasmids con- taining a HSV origin of DNA replication. This system is similar in concept to those described for the oriP system of Epstein-Barr virus (41, 42) and for bovine papilloma virus (43). Two observations support the idea that the replication observed in this system is an accurate model for HSV DNA replication. First, replication is completely dependent on the presence, in cis, of either one of the two HSV origins of DNA replication, oris or oriL; plasmids that lacked one of these sequences did not replicate. Second, replication depends on at least two of the genes known from conventional genetic analysis to be required for viral DNA replication: the genes encoding Pol and DBP (10-18). In addition to the plasmid containing both of these genes (pMC122), replication also requires the presence of four other plasmids. Thus, in this system, replication depends on the simultaneous expression of at least six genes, which are contained on at least five different cotransfected plasmids.
Some of the viral gene products required in this system may not be directly involved in DNA replication but may instead have regulatory roles. The data suggest that the immediate early protein IE175 is required for DNA replica- tion in this system: the only sequences present in EcoRI JK that are not also present within Xba I D encode IE175 (44). Genetic data indicate that this protein is required for the expression of early genes in infected cells (21, 22), and several groups have shown that a plasmid carrying this gene can activate the expression of a HSV early gene contained on
This content downloaded from 169.229.32.136 on Wed, 7 May 2014 19:09:24 PMAll use subject to JSTOR Terms and Conditions

9098 Genetics: Challberg Proc. Natl. Acad. Sci. USA 83 (1986)
a cotransfected plasmid (45-48). Thus, it seems likely that the requirement for EcoRI JK reflects a requirement for IE175 in the expression of genes directly involved in DNA replication, although a more direct role for IE175 in DNA replication cannot be eliminated. It remains to be seen whether other viral gene products are also required for the expression of early genes in this system.
The functional relationship between the two HSV origins, oris and oriL, is not known. The nucleotide sequences of the two origins share extensive homology (6, 33). oriL contains a sequence that comprises a perfect palindrome of 144 bp; ons contains a related sequence, but the extent of dyad symmetry is considerably less than in oriL. This difference between the two origins has prompted speculation that viral DNA syn- thesis originating at oriL is symmetric, whereas that originat- ing at oris is asymmetric (6, 33). In the system described in this paper, DNA replication on plasmids containing either oris or oriL can be assayed simultaneously (see Fig. 6, for example). Thus, further analysis should provide an answer to the question of whether there are any HSV genes that are specific for one of the two origins. From a practical view- point, the large region of perfect dyad symmetry within oriL causes marked instability in E. coli plasmids containing this region of the genome; most clones containing this region carry small deletions that reduce the extent of or eliminate symmetry (6, 33). The two subclones of Xba I F described in this paper, pNN1 and pNN3, both have an identical 150-bp deletion removing virtually all of the 144-bp palindrome in oriL. As shown by Weller et al. (6), this deletion completely inactivates origin function. Since the genes on either side of the palindrome, dbp and pol, are expressed at a level that is sufficient to support oris-dependent replication, it can be concluded that expression of these two genes is not com- pletely dependent on the presence of a functional origin in cis. The significance of the close proximity of oriL to the transcriptional start site of two genes directly involved in DNA replication (dbp and pol) remains unclear.
The genes which are known from genetic experiments to be involved in viral DNA replication are all contained in one of the five plasmids required for transient DNA replication. Thus, DBP and Pol are encoded in Xba I F (14, 18), RR is located in Xba I E (19), alkaline Exo is specified in Xba I C (49), and, as mentioned, IE175 is contained in EcoRI JK (21, 22). Since the plasmid containing Xba I D is also required for DNA replication in this system, there must be at least one additional gene necessary for DNA replication which has not yet been identified. It seems likely that some of the other plasmids contain other unidentified replication genes as well. Additional subcloning of each of the plasmids required in this system should allow one to identify all of the HSV genes required for DNA replication in cultured cells. Precise identification of the sequences encoding the required repli- cation functions should facilitate biochemical analysis of viral DNA replication. Finally, it seems clear that HSV DNA replication is a complex process involving the interaction of several different components. The work reported in this paper shows that a transient assay system involving the cotransfection of many plasmids can be used to study this multicomponent system. It seems likely that other complex systems for which suitable assays exist can be analyzed in similar fashion.
I thank Nancy Nelson for expert technical assistance and Drs. Andrew Davison and Thomas Kelly for stimulating discussions and for helpful comments on this manuscript. I am particularly grateful to Dr. Sandra Weller for virus stocks, plasmids, the encouragement to start on this work, and continuing advice. Finally, I thank Dr. Bernard Moss for support and encouragement.
1. Becker, Y., Dym, H. & Sarov, 1. (1968) Virology 36, 184-192. 2. Spaete, R. R. & Frenkel, N. (1982) Cell 30, 295-304. 3. Vlasny, D. A. & Frenkel, N. (1981) Proc. Natl. Acad. Sci. USA 78,
742-746. 4. Stow, N. D. (1982) EMBO J. 1, 863. 5. Stow, N. D. & McMonagle, E. C. (1983) Virology 130, 427-438. 6. Weller, S. L., Spadaro, A., Schaffer, J. E., Murray, A. W.,
Maxam, A. M. & Schaffer, P. A. (1985) Mol. Cell. Biol. 5, 930-942. 7. Jacob, R. J. & Roizman, B. (1977) J. Virol. 23, 394-411. 8. Jacob, R. J., Morse, L. S. & Roizman, B. (1979) J. Virol. 29,
448-457. 9. Jongeneel, C. V. & Bachenheimer, S. L. (1981) J. Virol. 39,
656-660. 10. Hay, J. & Subak-Sharpe, J. H. (1976) J. Gen. Virol. 31, 145-148. 11. Chartrand, P., Stow, N. D., Timbury, M. C. & WiLkie, N. M.
(1979) J. Virol. 31, 265-276. 12. Coen, D. M., Furman, P. A., Gelep, P. T. & Schaffer, P. A. (1982)
J. Virol. 41, 909-918. 13. Honess, R. W., Purifoy, D. J. M., Young, D., Gopal, R., Com-
mack, N. & O'Hare, P. (1984) J. Gen. Virol. 65, 1-17. 14. Coen, D. M., Aschman, D. P., Gelep, P. T., Retondo, M. J.,
Weller, S. K. & Schaffer, P. A. (1984) J. Virol. 49, 236-247. 15. Bayliss, G. J., Marsden, H. S. & Hay, J. (1975) Virology 68,
124-134. 16. Powell, K. L. & Purifoy, D. J. M. (1976) Intervirology 7, 225-239. 17. Powell, K. L., Littler, E. & Purifoy, D. J. M. (1981) J. Virol. 31,
145-148. 18. Weller, S. K., Lee, K. J., Sabourin, D. J. & Schaffer, P. A. (1983)
J. Virol. 45, 354-366. 19. Preston, V. G., Palfreyman, J. W. & Dutia, B. M. (1984) J. Gen.
Virol. 65, 1457-1466. 20. Moss, H. (1986) J. Gen. Virol. 67, 1173-1178. 21. Preston, C. M. (1979) J. Virol. 29, 275-284. 22. Dixon, R. A. F. & Schaffer, P. A. (1980) J. Virol. 36, 189-203. 23. Honess, R. W. & Roizman, B. (1974) J. Virol. 14, 8-19. 24. Honess, R. W. & Roizman, B. (1975) Proc. Natl. Acad. Sci. USA
72, 1276-1280. 25. Matz, B., Subak-Sharpe, J. H. & Preston, V. G. (1983) J. Gen.
Virol. 64, 2261-2270. 26. Chu, C.-T., Parris, D. S., Dixon, R. A. F., Farber, F. E. & Schaf-
fer, P. A. (1979) Virology 98, 169-181. 27. Spear, P. G. & Roizman, B. (1980) in Molecular Biology of DNA
Tumor Viruses, ed. Tooze, J. (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY), 2nd Ed., Part 2, pp. 615-745.
28. Wagner, E. K. (1985) in The Herpesviruses, ed. Roizman, B. (Plenum, New York), Vol. 3, pp. 45-104.
29. Denniston, K. J., Madden, M. J., Enquist, L. W. & Vande Woude, G. (1981) Gene 15, 365-378.
30. Birnboim, H. C. & Doly, J. (1979) Nucleic Acids Res. 7, 1513-1523.
31. Hanahan, D. (1983) J. Mol. Biol. 166, 557-580. 32. Yanisch-Perron, C., Vieira, J. & Messing, J. (1985) Gene 33,
103-119. 33. Quinn, J. P. & McGeoch, D. J. (1985) Nucleic Acids Res. 13,
8143-8163. 34. Gibbs, J. S., Chiou, H. C., Hall, J. D., Mount, D. W., Retondo,
M. 3., Weller, S. K. & Coen, D. M. (1985) Proc. Natl. Acad. Sci. USA 82, 7669-7673.
35. Graham, F. L. & Van der Eb, A. J. (1973) Virology 52, 456-467. 36. Graham, F. L. & Van der Eb, A. J. (1973) J. Virol. 38, 54-58. 37. Maniatis, T., Fritsh, E. F. & Sambrook, J. (1980) Molecular
Cloning. A Laboratory Manual (Cold Spring Harbor Laboratories, Cold Spring Harbor, NY).
38. Peden, K. W. C., Pipas, J. M., Pearson-White, S. & Nathans, D. (1980) Science 209, 1392-1396.
39. Goldin, A. L., Sandri-Goldin, R. M., Levine, M. & Glorioso, J. C. (1981) J. Virol. 38, 50-58.
40. Skare, J. & Summers, W. C. (1977) Virology 76, 581-595. 41. Yates, J. L., Warren, N. & Sugden, B. (1985) Nature (London)
313, 812-815. 42. Lupton, S. & Levine, A. J. (1985) Mol. Cell. Biol. 5, 2533-2542. 43. Lusky, M. & Botchan, M. R. (1986) Proc. Natl. Acad. Sci. USA
83, 3609-3613. 44. Murchie, M. J. & McGeoch, D. J. (1982) J. Gen. Virol. 62, 1-15. 45. Everet, R. D. (1984) EMBO J. 3, 3135-3141. 46. O'Hare, P. & Hayward, G. S. (1985) J. Virol. 53, 751-760. 47. Quinlan, M. P. & Knipe, D. M. (1985) Mol. Cell. Biol. 5, 957-963. 48. DeLuca, N. A. & Schafer, P. A. (1985) Mol. Cell. Biol. 5,
1997-2008. 49. Preston, C. M. & Cordingly, M. G. (1982) J. Virol. 43, 386-394.
This content downloaded from 169.229.32.136 on Wed, 7 May 2014 19:09:24 PMAll use subject to JSTOR Terms and Conditions