486 immunoogic disorders
TRANSCRIPT

Immunologic Disorders
RESOURCE FACULTYDr. Jyotsna Rimal
Additional professor and HOD
Dr. Iccha kumar Maharjan
Associate professor
DEPARTMENT OF ORAL MEDICINE AND RADIOLOGY PRESENTER
Nabin Chaudhary
BDS 2011

Contents Immunodeficiency Primary immunodeficiencies
Deficiencies in innate immunity:phagocyte deficiencies
Deficiencies in adaptive immunity
T-cell deficiencies
B-cell deficiencies Secondary immunodeficiencies
Innate immune system Autoimmune disease
SLE
Scleroderma
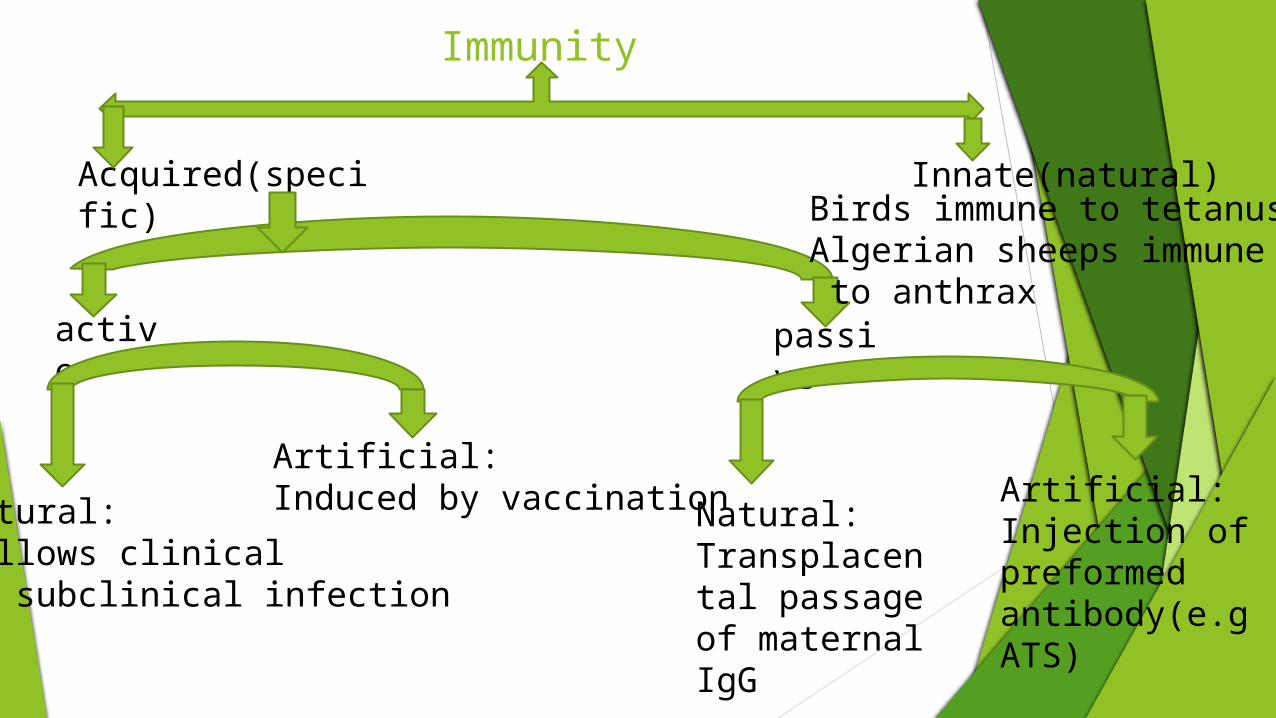
Immunity
Acquired(specific) Innate(natural)
active passive
Natural:Follows clinical or subclinical infection
Artificial:Induced by vaccination Natural:
Transplacental passageof maternal IgG
Artificial:Injection of preformed antibody(e.g ATS)
Birds immune to tetanus;Algerian sheeps immune to anthrax

INNATE IMMUNITY Barriers
Physical
skinhairmucous
Chemicalsweat tearssalivastomach acidurine

Cells of innate immunity
Cells Location Function
Monocytes Blood Phagocytosis
Macrophages Tissue Phagocytosis, inflamation, Antigen presentation
Neutrophils Blood Phagocytosis
Eosinophils Tissue/blood Cytolysis
Basophils Blood Inflammation
Mast cells Tissue Inflammation
Natural killer cells Blood/Tissue Cytolysis
Dendritic cells Tissue Antigen presentation

Adaptive immune response
Third line of defense Precisely adapted to each specific microorganism encountered by an individual Memory Mounts rapid and efficient response in the next encounter Consists of Cells:
B lymphocytes T lymphocytes
Molecules: Antibodies, Cytokines
Primary:Thymus Bone marrow
Secondary:Payer’s patchSpleen Lymph node
ORGANS
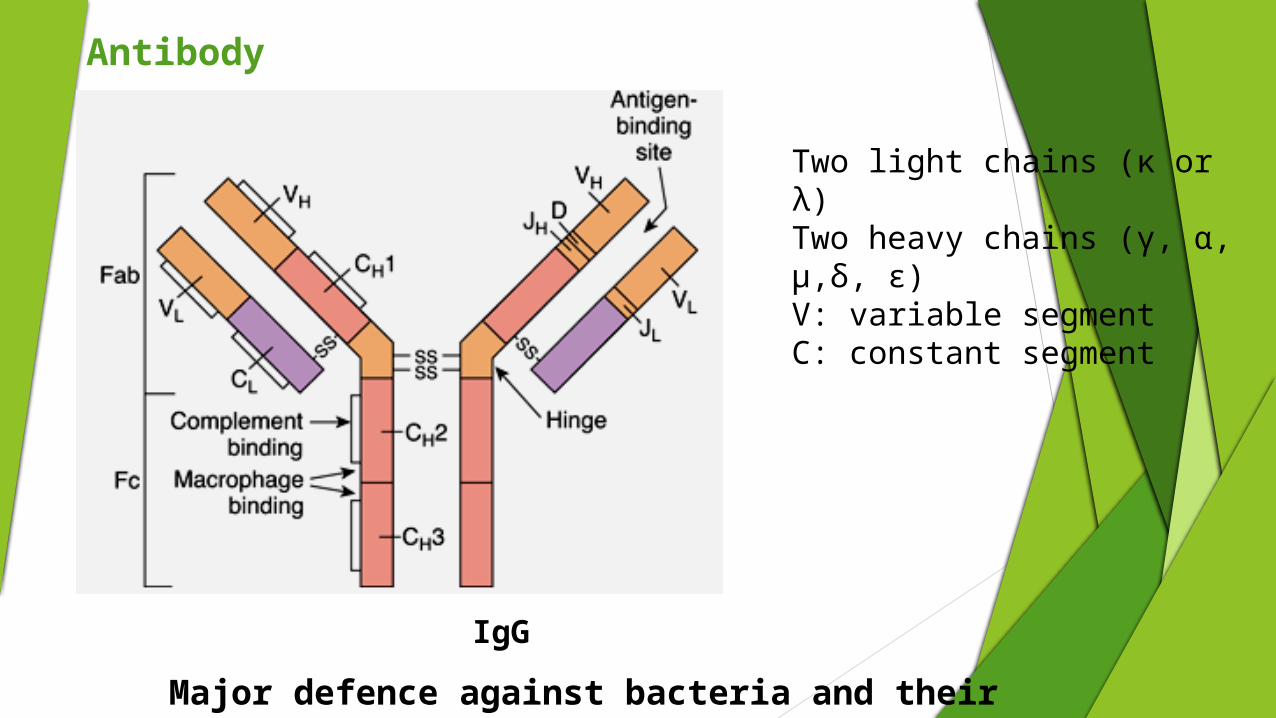
Antibody
Two light chains (κ or λ)Two heavy chains (γ, α, μ,δ, ε)V: variable segmentC: constant segment
IgG
Major defence against bacteria and their toxin

% of total immuno-globulin
Functions
IgG 80 Complement activation, Major defence against bacteria and their toxin
IgA 13 Localized protection in external secretions (tears, intestinal secretions, etc)
IgM 6 Complement activation, Antigen recognition by B cells
IgD 0.1 Antigen recognition by B cells
IgE 0.002 Major defence against helminths; mediates allergic reation, releases histamine from basophils and mast cells
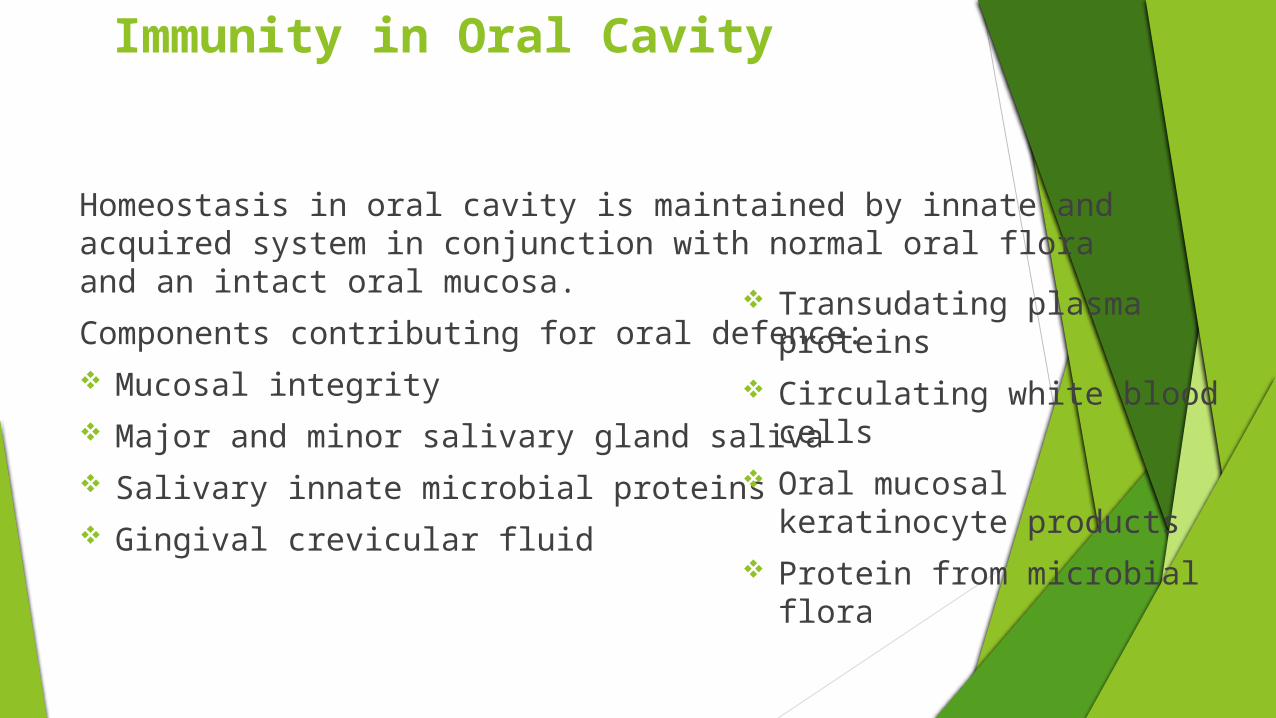
Immunity in Oral Cavity
Homeostasis in oral cavity is maintained by innate and acquired system in conjunction with normal oral flora and an intact oral mucosa.
Components contributing for oral defence: Mucosal integrity Major and minor salivary gland saliva Salivary innate microbial proteins Gingival crevicular fluid
Transudating plasma proteins
Circulating white blood cells
Oral mucosal keratinocyte products
Protein from microbial flora

IMMUNODEFICIES
PRIMARY IMMUNODEFECIENCY(<10%)• TCELL DEFECT• B CELL DEFECT• COMPLEMENT DEFECT• PHAGOCYTIC DEFECT
SECONDARY IMMUNODEFECIENCY(90%).DRUGS.INFECTIONS
Congenital Manifests since early age
Acquired Manifests at any ageResult either from production or destruction of immune cells orloss of components of humoral immunity

Immunodeficiency
T-cell deficiencies B-cell deficiencies
in T-cell number e.g.DiGeorge Syndrome(Velocardiofacial syndrome)
Qualitative T-cell defects e.g.Defects in MHC
in B-cell number e.g.Bruton’s X-linked Agammaglobulinemia and Non Bruton’s Agammaglobulinemia
in certain classes of immunoglobulins e.g. common variable immunodeficiency; selective immunoblobulin deficiency
Severe combined immunodeficiency(SCID)Wiskott Aldrich syndrome
Ataxia Telangiectasia

Oral complication with specific immunodeficiencies
Immune defect Disease example Common infectious susceptibility
Oral complications
Severe in neutrophil no.
Kostmann’s syndrome,Severe congenital neutropenia
Bacterial andFungal infections
Aggressive periodontitis,Recurrent oral ulcerations,Delayed wound healing,Candidiasis
Defective chemotaxis of neutrophils
Leukocyte adhesion deficiency
Bacterial andFungal infections
Aggressive periodontitis,Recurrent oral ulcerations,Delayed wound healing,candidiasis

Unknown Job’s syndrome Recurrent skin and mucous membrane infections
Delayed exfoliation of primary teeth,midline defects of tongue,oral candidiasis,characteristic face with broad nasal bridge
Severely T-and B-cell no.
SCID Infections of all types
Oral candidiasis,herpes,recurrent oral ulcerations,severe necrotizing gingival ulcerations
in T-cell no. DiGeorge syndrome
Recurrent viral and fungal infections
candidiasis,recurrent herpetic infections

Abnormal T-cell function
MHC class I deficiency of CD8 T-cellMHC class II deficiency of CD4 T-cell
Recurrent viral and fungal infections
candidiasis,recurrent herpetic infections
Severly B-cell no.
Bruton’s X-linked agammaglobulinemia
Recurrent respiratory and sinus infections
Possible sepsis from odontogenic infections
Solitary immunoglobulin class deficiency
Selective IgA deficiencySelective IgG deficiency
Varied from no symptoms to recurrent respiratory and sinus infections
Candidiasis,oral ulcerations,possible sepsis from odontogenic infections
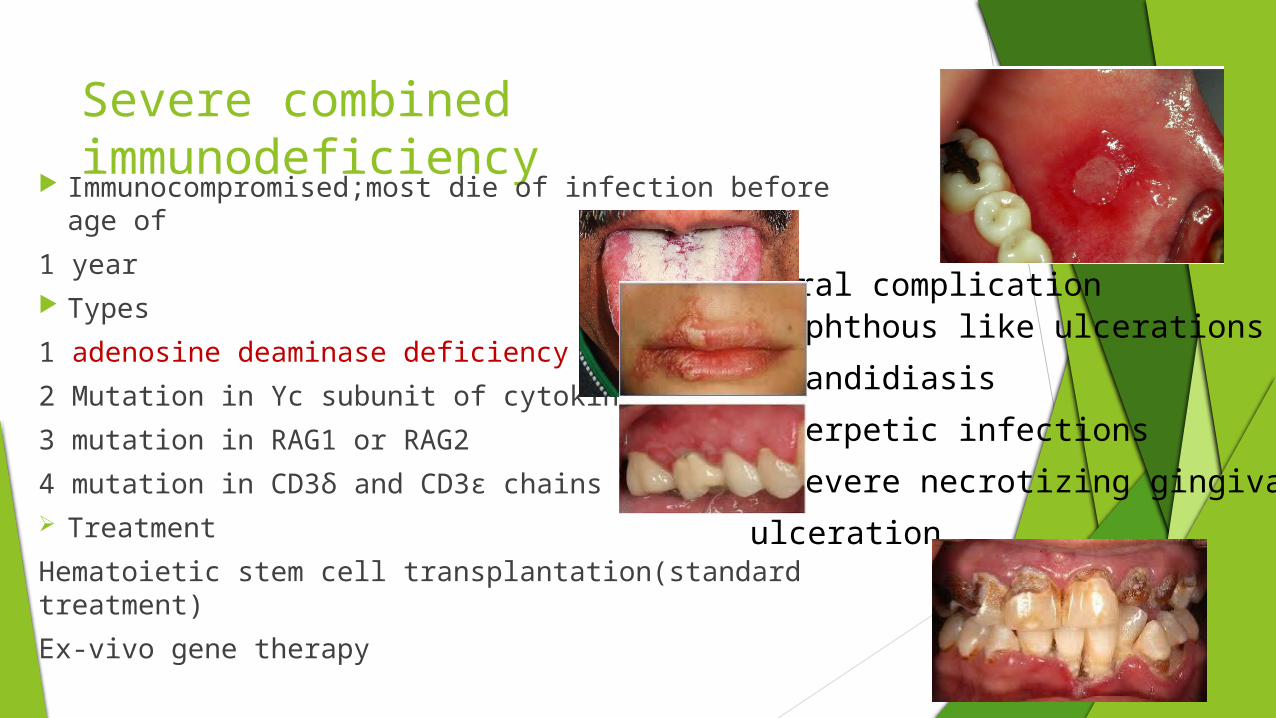
Severe combined immunodeficiency
Immunocompromised;most die of infection before age of
1 year Types
1 adenosine deaminase deficiency
2 Mutation in Υc subunit of cytokine receptors
3 mutation in RAG1 or RAG2
4 mutation in CD3δ and CD3ε chains Treatment
Hematoietic stem cell transplantation(standard treatment)
Ex-vivo gene therapy
Aphthous like ulcerations Candidiasis Herpetic infections Severe necrotizing gingival
ulceration
Oral complication
Prof Ariyanto Harsono MD PhD SpA(K) 16
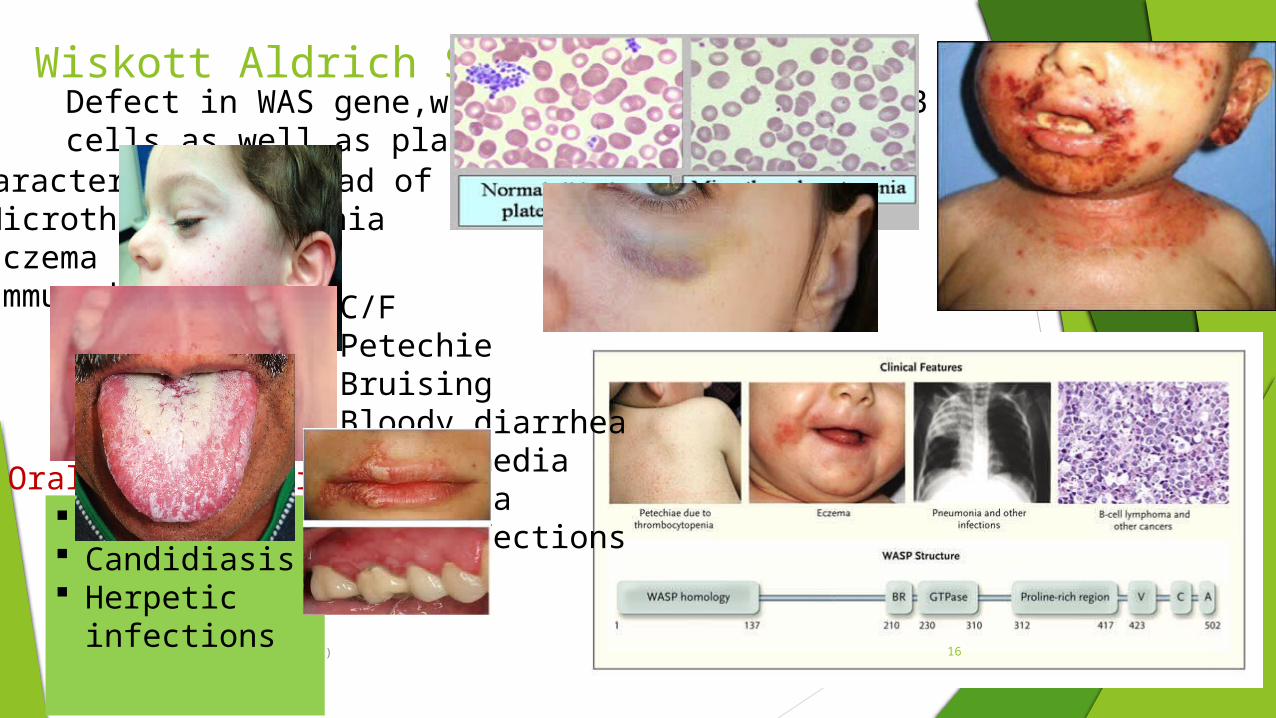
Wiskott Aldrich Syndrome
Oral manifestations Purpura Candidiasis Herpetic
infections
Characterised by triad of symptoms Microthrombocytopenia Eczema Immunodeficiency
Defect in WAS gene,which affects both T and B cells as well as platelets
C/FPetechie BruisingBloody diarrheaOtitis mediaPneumoniaSkin infections
17
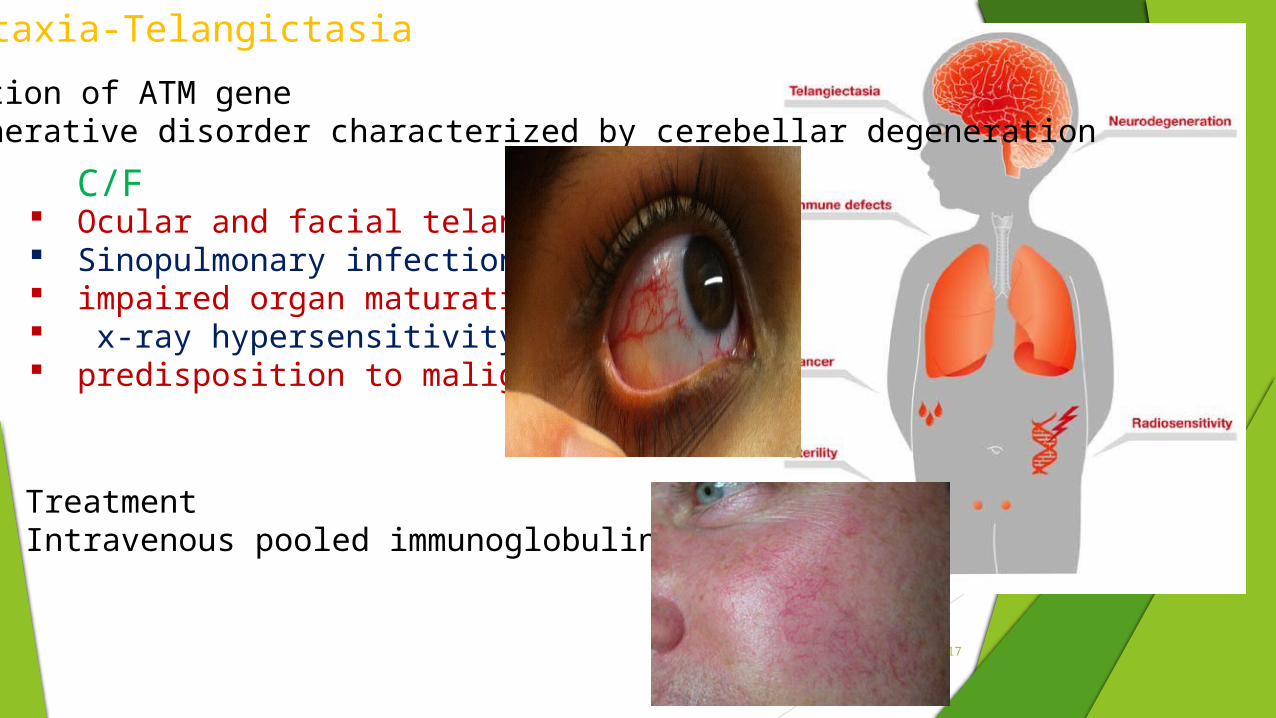
Ocular and facial telangictases Sinopulmonary infections impaired organ maturation x-ray hypersensitivity predisposition to malignancy.
Ataxia-Telangictasia
C/F
Mutation of ATM geneDegenerative disorder characterized by cerebellar degeneration
Treatment Intravenous pooled immunoglobulin
18
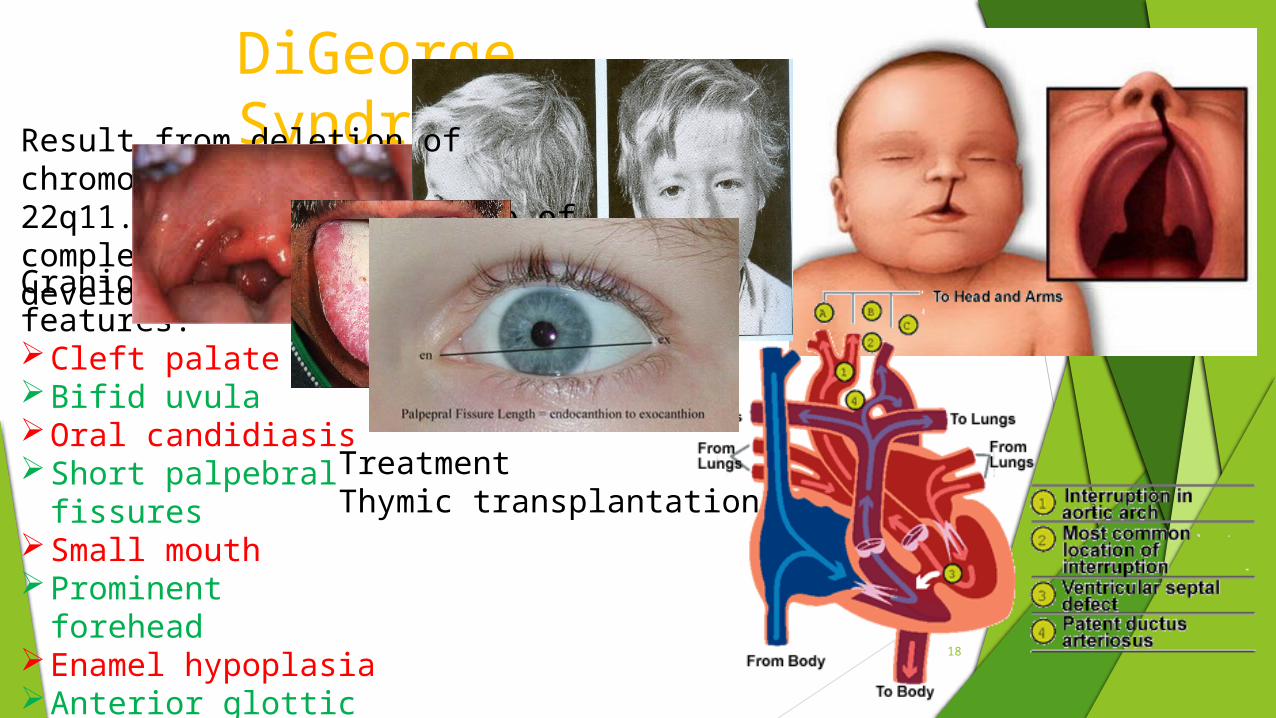
DiGeorge Syndrome
Craniofacial features:Cleft palateBifid uvulaOral candidiasisShort palpebral fissuresSmall mouthProminent foreheadEnamel hypoplasiaAnterior glottic webs
Result from deletion of chromosome22q11.2;leading to failure of completedevelopment of thymus
TreatmentThymic transplantation

X-linked agammaglobulinemia(bruton’s Disease)
Mutation in the gene called bruton thyroid kinase(BTK)Virtual absence of B-cellsVery low level of all immunoglobulins( IgG, IgA, IgM, IgD and IgE)
Treatment
Aggressive antibiotic treatment and intravenous pooled immunoglobulin transfusions

Secondary immunodeficiencies
Cellular(primarily neutrophils) Neutropenia results during cancer chemotherapy, stem cell transplantation,aplastic
anemia and autoimmune neutropenia If ANC<1000cells/mm3 risk of nfection.Thus antibiotic prophylaxis should be given
prior to invasive dental procedures Complement Acquired deficiency seen in advanced liver disease Associted with increased incidence of bacterial infections and malignancies(CLL)
Innate immune system

Adaptive immune system
Decrease in T-cell no eg HIV and/or function eg chronic GVH disease,treatment with anti tumor necrosis(TNF-α)
Agents Infections with intracellular bacteria and fungi(pneumocystis
carinii),viruses(herpes family,papilloma virus) and parasites(Toxoplasma and cryptosporidium) are typical of T-cell-deficient states
Oral candidiasis(advanced HIV) Herpes zoster(CLL)
Homoral(antibodies)
• Associated with increased loss of immunoglobulin eg nephrotic syndrome• And decreased production of immunoglobulin eg multiple myeloma, CLL
Cell mediated

Case study Immunological investigations
Quantitative serum immunoglobulins (g/L):
IgG 0.17 (5.5–10.0)
IgA Not detected (0.3–0.8)
IgM 0.07 (0.4–1.8)
Antibody activity
Immunization responses – no detectable IgG antibodies to:
Tetanus toxoid (post Imx)
Haemophilus type b polysaccharides (post Imx)
Polio (post Imx)
Measles (post Imx)
Rubella (post Imx)
Isohaemagglutinins (IgM) not detected (blood group A Rh+)
Blood lymphocyte subpopulations (×109/L):
Total lymphocyte count 3.5 NR* (2.5–5.0)
T lymphocytes (CD3) 3.02 NR (1.5–3.0)
B lymphocytes (CD19) <0.1 NR (0.3–1.0)
Peter was born after an uneventful pregnancy. At 3 months, he developed otitis media At the ages of 5 months and 11 months, he
was admitted to hospital with untypable Haemophilus influenzae pneumonia.
He is the fourth child of unrelated parents;his three sisters showed no predisposition to infection.
All immunizations were uneventful. severe reduction in all three classes
of serum immunoglobulins and no specific antibody production
mutation in the Btk gene was detected

Diagnosis???
Bruton’s disease(X-linked agammaglobulinaemia) Treated by 2-weekly intravenous infusions of
human normal IgG in a dose of 400 mg/kg body weight/month.

McQ Which of the following Ig is involved in mediating allergic reactions a) IgG b) IgM c) IgE d) IgA
All of the following are autoimmune disorders except a) Graves disease b) SCID c) Rheumatoid arthritis d) Addison’s disease
(b)
(c)

Systemic lupus erythematosus
Autoimmune disorder characterized by multisystem microvascular inflammation with the generation of autoantibodies
When the skin is involved- lupus dermatitis or cutaneous lupus erythematosus.
isolated to the skin, without internal disease- discoid lupus.
internal organs are involved-systemic lupus erythematosus (SLE)
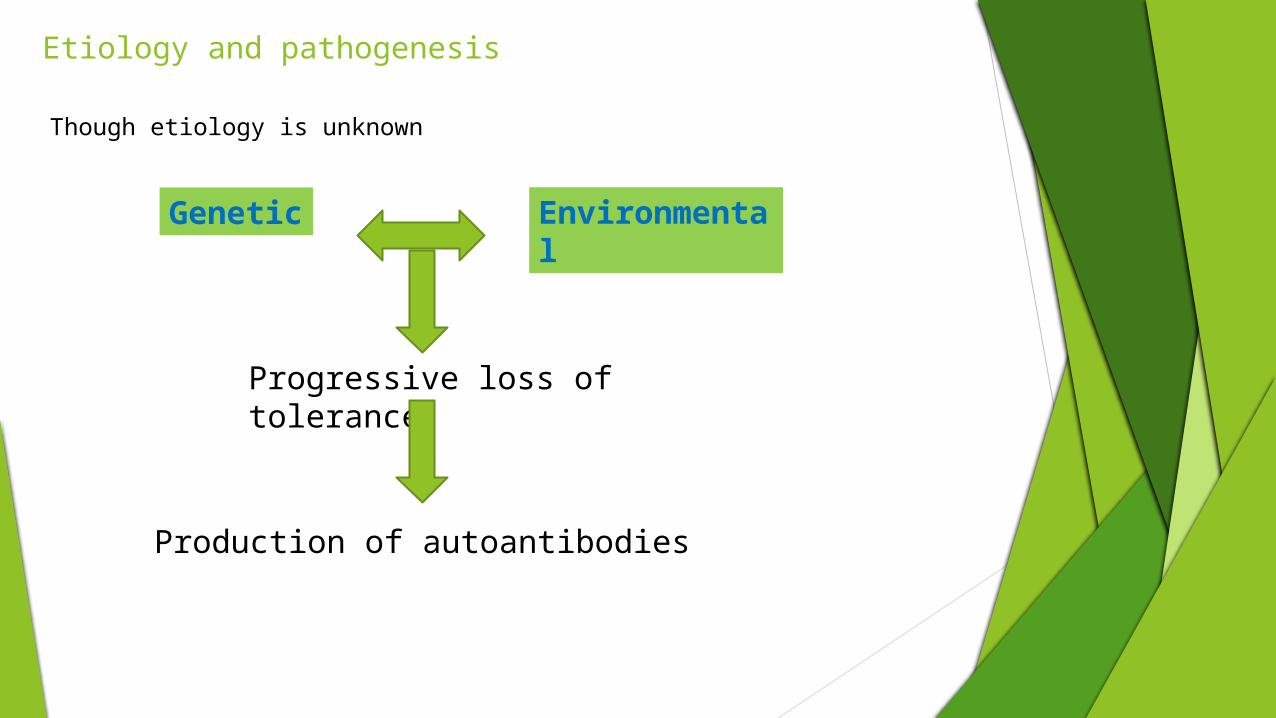
Etiology and pathogenesis ____________________
Genetic Environmental
Though etiology is unknown
Progressive loss of tolerance
Production of autoantibodies
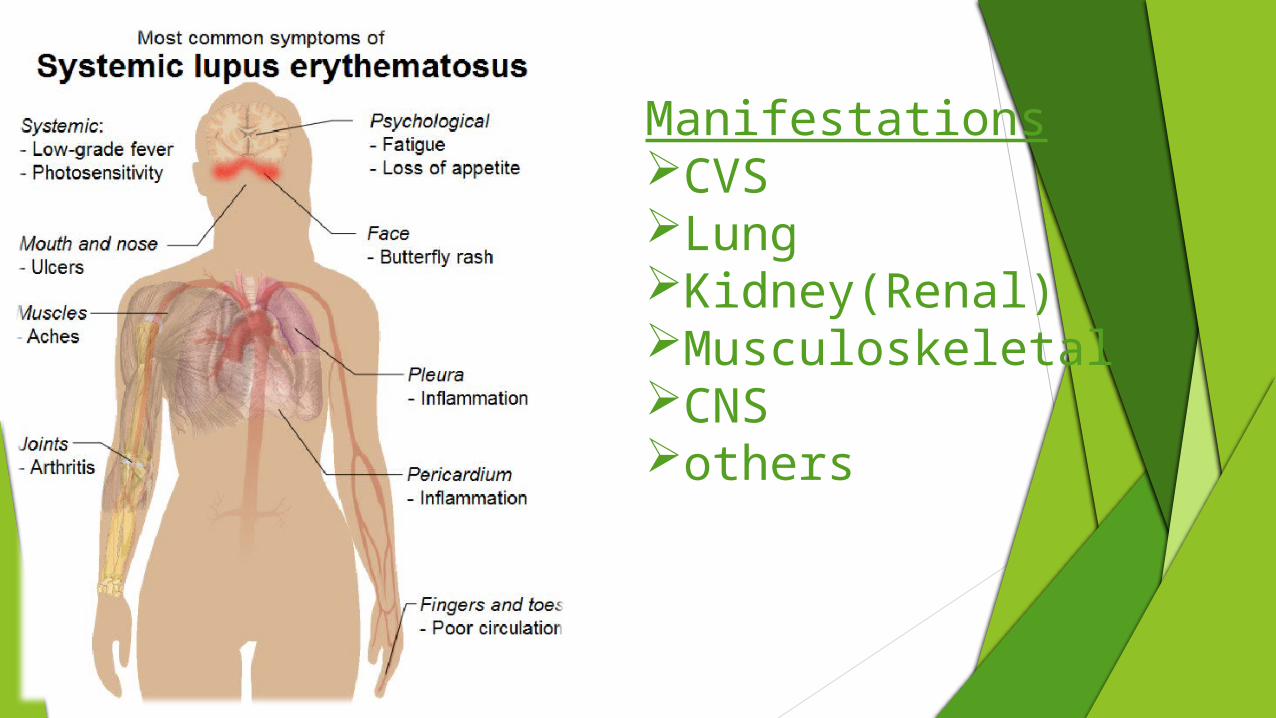
ManifestationsCVSLungKidney(Renal)MusculoskeletalCNSothers

American college of Rheumatology criteria
MALAR RASH
DISCOID RASH
PHOTOSENSITIVITY
ORAL ULCERS
ARTHRITIS
SEROSITIS
RENAL DISORDER
NEUROLOGIC DISORDER
HEMATOLOGICAL DISORDER
IMMUNOLOGIC DISORDER
ANTINUCLEAR ANTIBODY
If ≥4 of these criteria,are present diagnosis likely to be SLE ;sensentivity=70-96%;specificity=90-100%
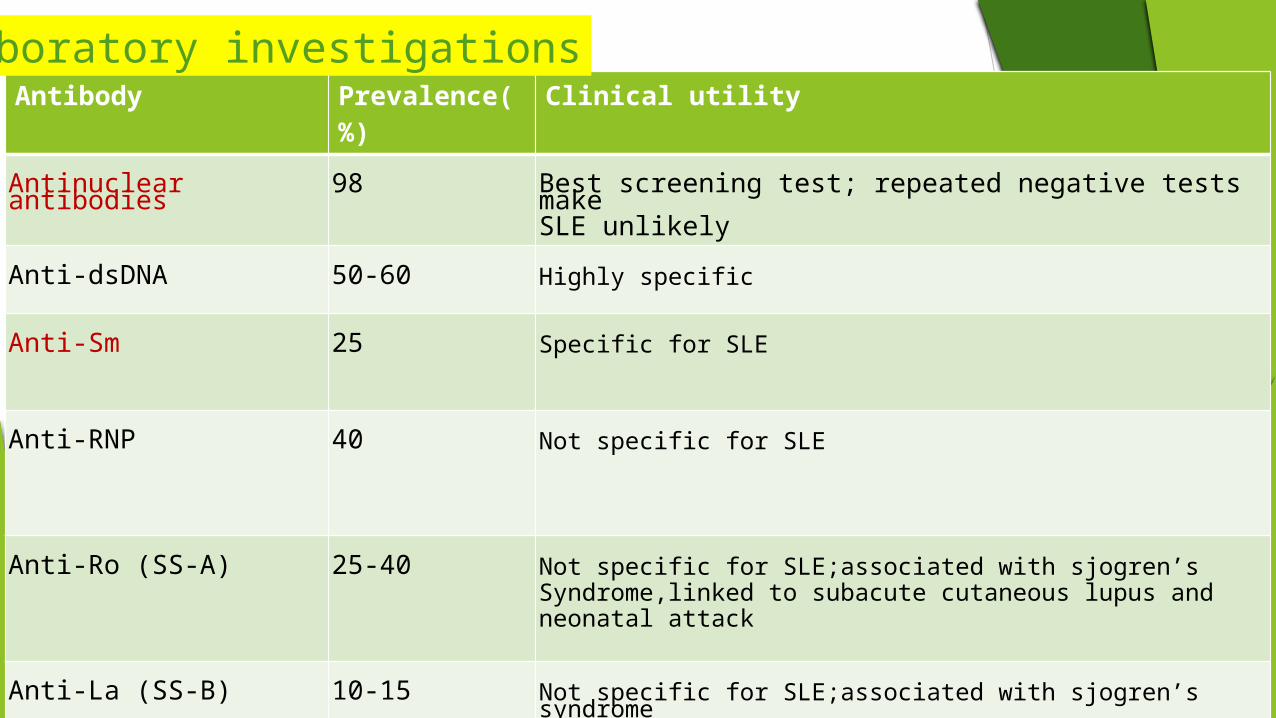
Antibody Prevalence(%) Clinical utility
Antinuclear antibodies 98 Best screening test; repeated negative tests make SLE unlikely
Anti-dsDNA 50-60 Highly specific
Anti-Sm 25 Specific for SLE
Anti-RNP 40 Not specific for SLE
Anti-Ro (SS-A) 25-40 Not specific for SLE;associated with sjogren’s Syndrome,linked to subacute cutaneous lupus and neonatal attack
Anti-La (SS-B) 10-15 Not specific for SLE;associated with sjogren’s syndrome
Laboratory investigations

Antihistone 70 More frequent in drug-induced lupus than in SLE
Antiphospholipid 50Three tests available— ELISA s for cardiolipin and 2G1, sensitive prothrombin time (DRVVT); predisposes to clotting, fetal loss, thrombocytopenia
Antierythrocyte 60 Measured as direct Coombs' test; a small proportion develops overt hemolysis
Antiplatelet 30 Associated with thrombocytopenia but sensitivity and specificity are not good; this is not a useful clinical test
Antineuronal (includes anti-glutamate receptor)
60 In some series a positive test in CSF correlates with active CNS lupus
Antiribosomal P 20 correlates with depression or psychosis due to CNS lupus

Immunohistology:
Direct Immunofluorescence
Lupus band test:
-Deposition of IgG;IgM and C3 in a band like pattern at the dermo-epidermal junction.
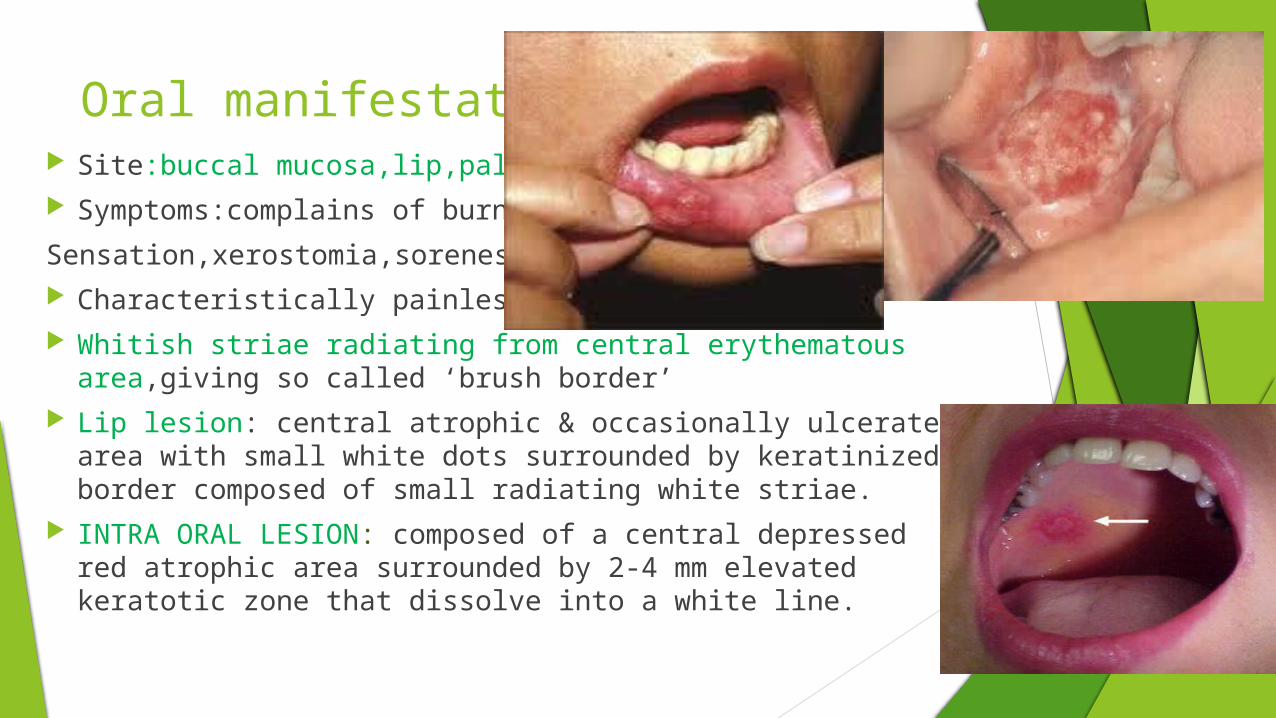
Oral manifestations Site:buccal mucosa,lip,palate,gingiva Symptoms:complains of burning
Sensation,xerostomia,soreness of mouth Characteristically painless Whitish striae radiating from central erythematous area,giving so
called ‘brush border’ Lip lesion: central atrophic & occasionally ulcerated area
with small white dots surrounded by keratinized border composed of small radiating white striae.
INTRA ORAL LESION: composed of a central depressed red atrophic area surrounded by 2-4 mm elevated keratotic zone that dissolve into a white line.

Dental Management Risk of infection:
• Complete blood count
• If ANC<1000 cells/mm3;elective oral surgical procedures with potential for bacteremia should be delayed
• Antibiotic prophylaxis often recommended
Risk of bleeding
Platelet transfusions recommended in surgical patients with platelet count<50,000/mm3
controlled by local measures such as pressure and application of hemostatic agents,
platelet transfusion can be performed postoperatively if bleeding occurs despite local measures
Oral surgical procedures are safe with INR=2-3.5 and do not require discontinuation of anticoagulants
Adrenal suppression
Any patients with supraphysiologic corticosteroids intake for 2 weeks or longer in 2years time is at risk of adrenal suppression

Adrenal insufficiency:corticosteroid supplementation
Dental surgical procedures Previous systemic steroid use Current systemic steroid use
Minor oral surgery lasting <1 hr with local anesthetic only
Consider supplementation with 25mg of hydrocortisone equivalent before surgery;esp. if patient is infected
Consider supplementation with 25mg of hydrocortisone equivalent before surgery;esp. if patient is infected.No need to supplement if the patient is taking over 50mg hydrocortisone equivalent daily
Oral surgery with or without GA lasting >1 hr
Major oral surgery with GA lasting >1 hr and having significant blood loss,such as cancer or orthognathic surgery
50-100mg of hydrocortisone equivalent the day of surgery,with continuation of the dose for an additional day on the amount of
blood loss,presence of infection and length of surgery
Usual daily dose before surgery and hydrocortisone equivalent of 5o mg hydrocortisone IV during surgey and every 8hr after initial
dose for upto 72hr
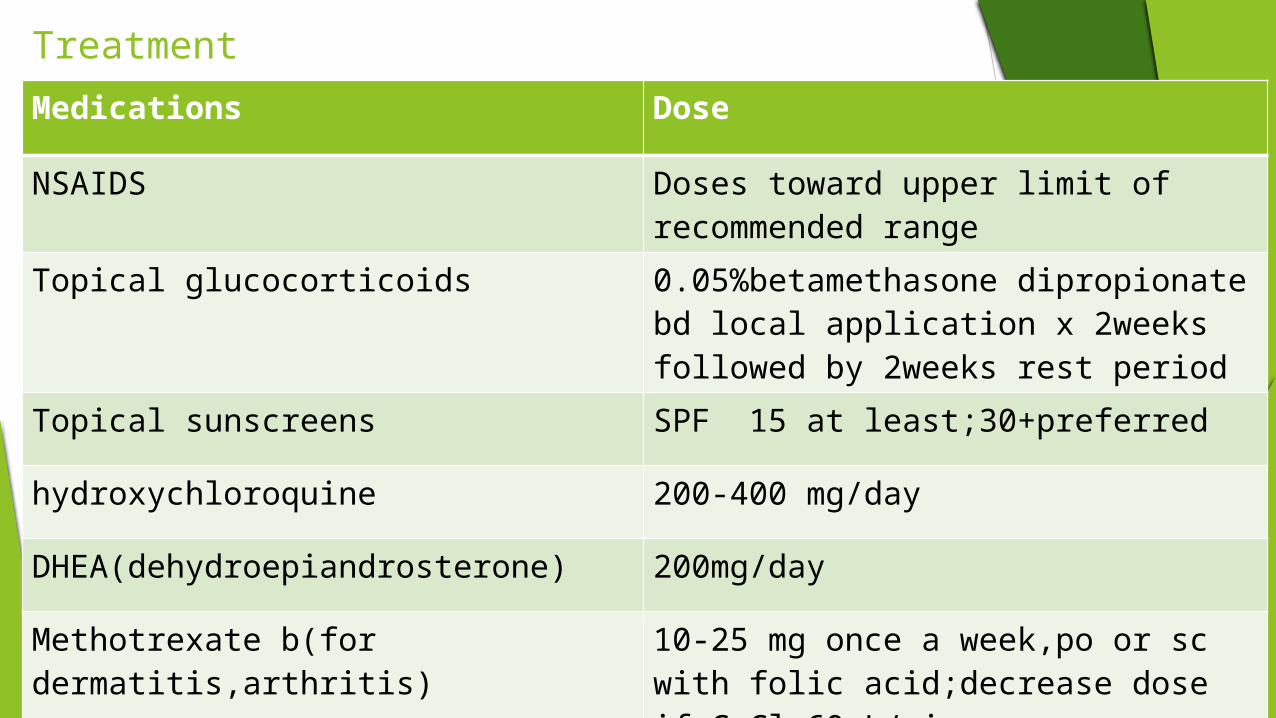
Treatment Medications Dose
NSAIDS Doses toward upper limit of recommended range
Topical glucocorticoids 0.05%betamethasone dipropionate bd local application x 2weeks followed by 2weeks rest period
Topical sunscreens SPF 15 at least;30+preferred
hydroxychloroquine 200-400 mg/day
DHEA(dehydroepiandrosterone) 200mg/day
Methotrexate b(for dermatitis,arthritis) 10-25 mg once a week,po or sc with folic acid;decrease dose if CrCl<60mL/min

Glucocorticoids oral Prednisolone:0.5-1 mg/kg per day for severe SLE0.07-0.3 mg/kg per day or qod for mild SLE
Methylprednisolone sodium succinate iv For severe dz;1g iv/day x 3days
cyclophosphamide IV 7-25 mg/kg /month x 6month;consider mesna administration with dose;1.5-3 mg/kg per day(decrease dose for CrCl<25ml/min)
Mycophenolate mofetil 2-3g/d PO(decrease dose if CrCl<25mL/min)
Azathioprine 2-3mg/kg per day PO(decrease frequency of dose if CrCl<50mL/min)

Scleroderma
Chronic disease characterised by
- thickening and fibrosis of skin Systemic sclerosis:fibrosis extends to internal
organs including heart,lungs,kidney,and GI tractTypes A) localized form i) linear ii) morphea B) progressive systemic sclerosis i) limited cutaneous scleroderma ii) diffuse cutaneous scleroderma
En coup de sabre
Morphea

Etiology and pathogenesis
Etiology is unclear Genetic predisposition Environmental exposure to pesticides,benzene derivatives,silica Viral triggers such as parvovirus B19 Pathogenesis is characterized by vascular damage Unregulated collagen deposition is a hallmark of disease

Clinical features
Raynaud’s phenomenon Cutaneous manifestations Musculoskeletal manifestations GI manifestations Cardiac manifestations Pulmonary manifestations Renal manifestations
Lab investigations• ANA(>90%patients)• Anticentromere(ACA)• AntitopoisomeraseI(Anti topo I)• Anti RNA polymerase I/III• Anti Th/To2-36
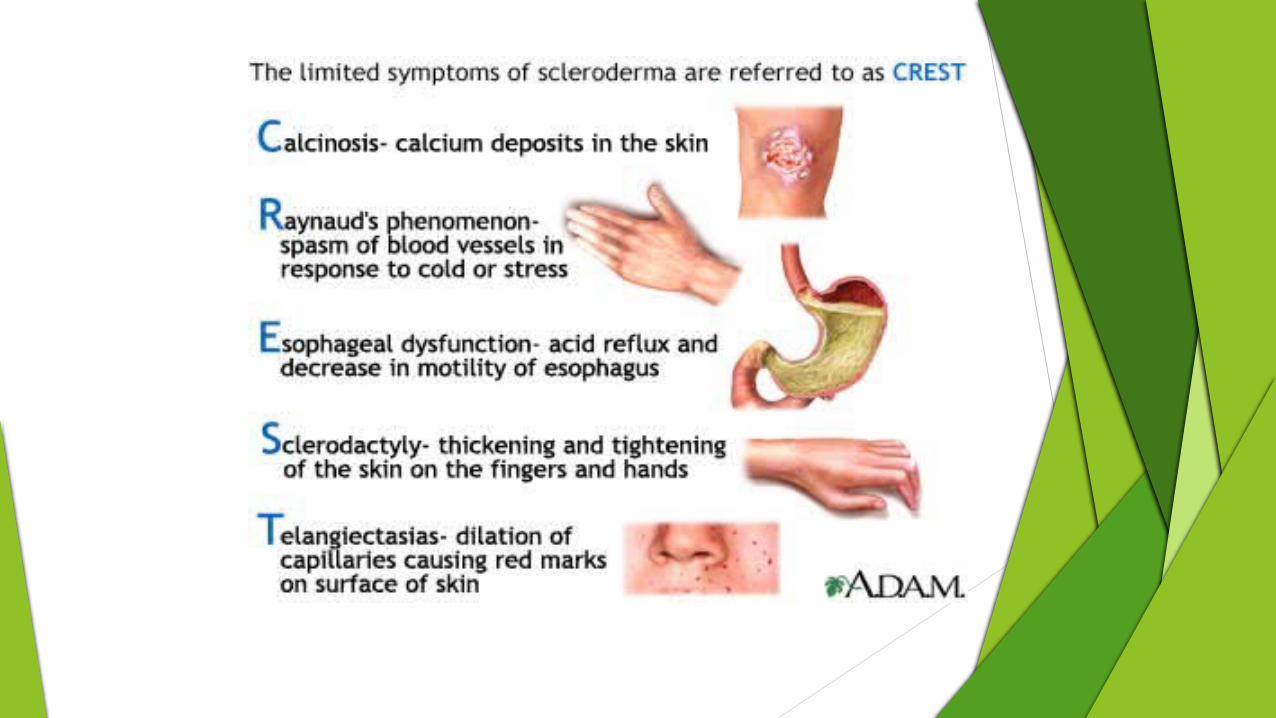
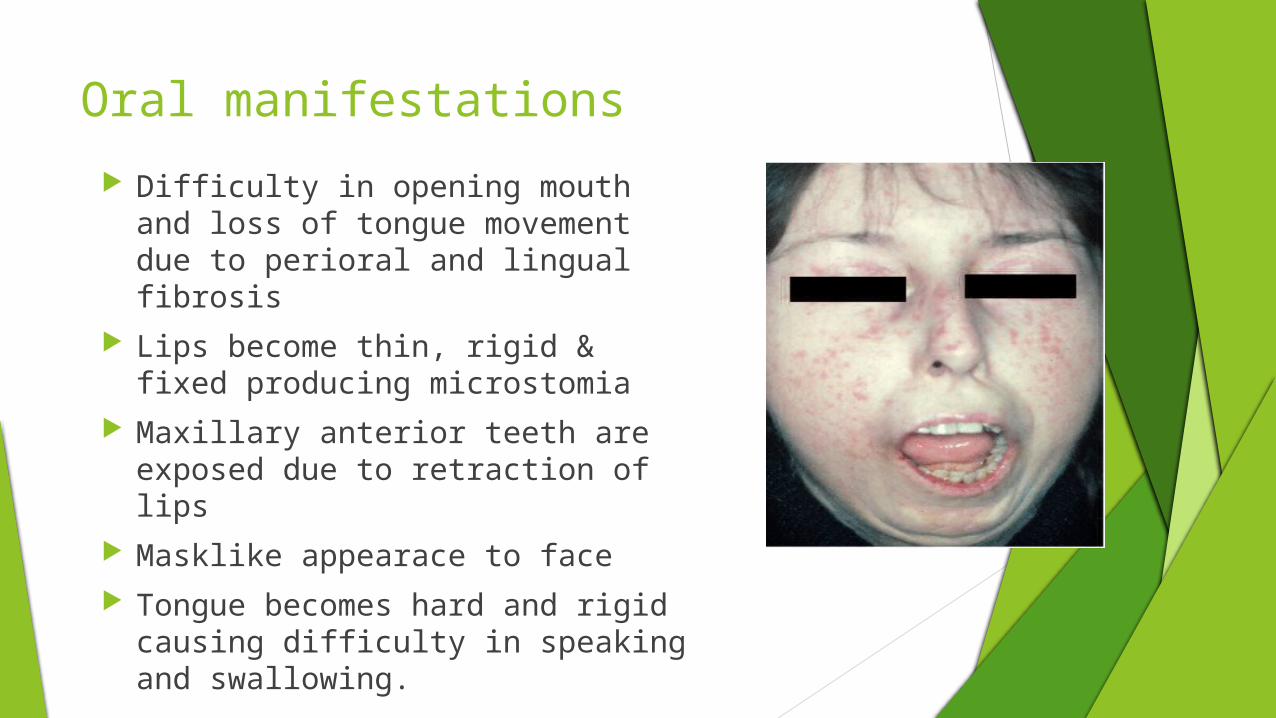
Oral manifestations
Difficulty in opening mouth and loss of tongue movement due to perioral and lingual fibrosis
Lips become thin, rigid & fixed producing microstomia
Maxillary anterior teeth are exposed due to retraction of lips
Masklike appearace to face Tongue becomes hard and rigid
causing difficulty in speaking and swallowing.
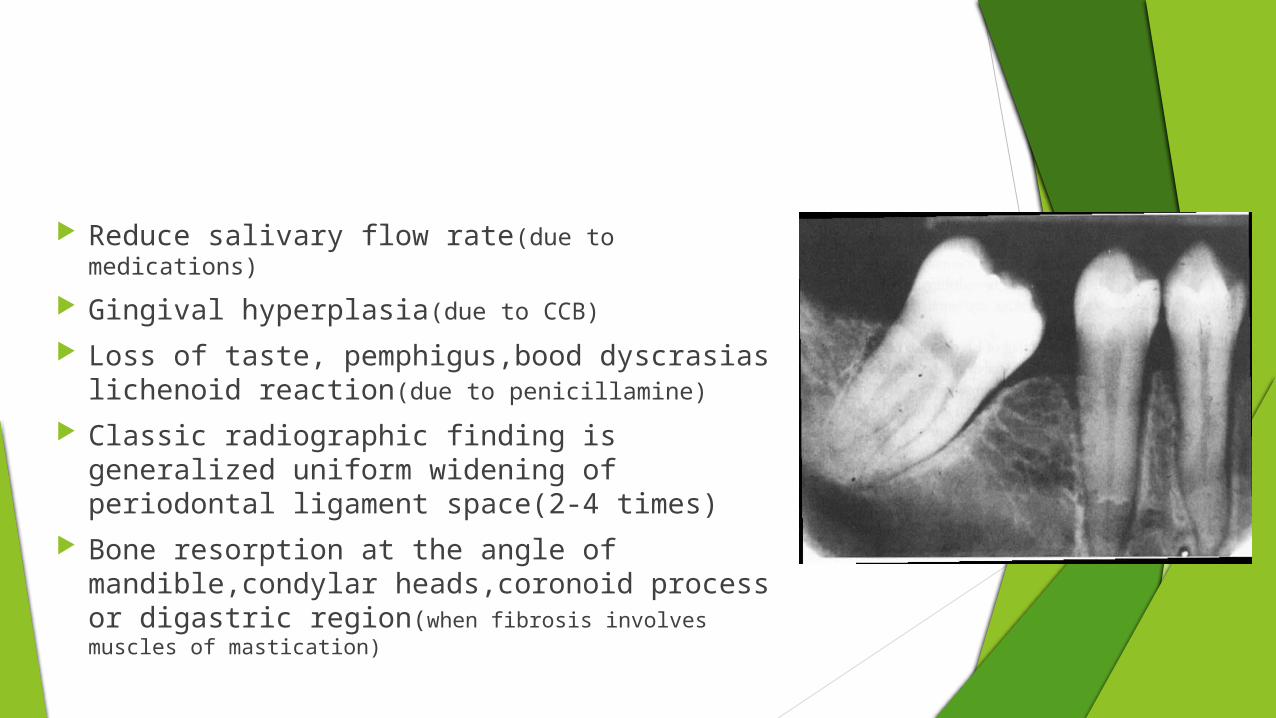
Reduce salivary flow rate(due to medications)
Gingival hyperplasia(due to CCB)
Loss of taste, pemphigus,bood dyscrasias lichenoid reaction(due to penicillamine)
Classic radiographic finding is generalized uniform widening of periodontal ligament space(2-4 times)
Bone resorption at the angle of mandible,condylar heads,coronoid process or digastric region(when fibrosis involves muscles of mastication)

Dental aspect Physical limitation caused by narrowing of oral aperture and
rigidity of tongue Poor oral hygiene;caries and plaque associated with decreasing
dexterity Dental procedures(molar endodontics,prosthetics,restoration in
posterior regions)become difficult Dental treatment altered due to physical problem of access Mouth stretch exercises No. of tongue blades increased between posterior teeth to
stretch facial muscles B/L commissurotomy

management
Non-pharmacological:
- Take warm water bath
- Avoid cold contact ( Especially in winters)
- Wear woolen gloves and socks
- Smoking and drugs like B-blocker inducing vasospasm is strictly prohibited

Pharmacological CCB, Nifedipine(30-90mg/day), non responsive once can use
Nicardipine(30-60mgbid) Combination of Pentoxifylline(400mg bid) and low dose aspirin(75
mg/day). Pentoxifylline improves digital perfusion by increasing the deformability of RBC plasma membrane
Immunomodulators: cyclosporine, methotrexate.
Antifibrotic therapy: D-PENICILLAMINE(start with 125-250mg OD then 250mg BD)
INFά & INFγ.

Case studyA 26-year-old woman presented with fatigue and painful, stiff knees of 4 weeks’ duration. She had a 6-year history
of raynaud’s phenomenon, frequent mouth ulcers and had had a blotchy rash
Bilateral effusions in both knee joints, but all other joints were normal.
She had no skin lesions, muscle tenderness, proteinuria or fever.
Full blood count showed mild thrombocytopenia and lymphopenia
InvestigationsC-reactive protein 8 mg/l (normal)Rheumatoid factor NegativeAntinuclear antibody PositiveAnti dsDNA-antibodies 80 (<25)
Serum complement levels C3 0.35 g/l (NR 0.65–1.30) C4 0.05 g/l (NR 0.20–0.50)
Serum immunoglobulins IgG 22.0 g/l (NR 7.2–19.0) IgA 3.8 g/l (NR 2.0–5.0) IgM 1.2 g/l (NR 0.5–2.0)Biopsy of normal, sun-exposed skin (lupus band test) Granular deposits of IgG and complement at dermo–epidermal junction Lupus band test
Well-demarcated digital ischaemia due to Raynaud’s phenomenon
Blotchy erythema on the dorsum of the hands

Diagnosis???
SLE treated with aspirin for her painful knees Following treatment with hydroxychloroquine, the
skin manifestations gradually disappeared

Revision
American college of rheumatology criteria for SLE Corticosteroids supplementation Most sensitive diagnostic test for SLE= CREST syndrome Most common problem associated with scleroderma for
dental procedures= Longest life which immunoglobulin= Immunoglobulin in secretions=
ANA
Trismus
IgG
IgA

McQ
In severe combined immune deficiency (SCID), the patients are deficient in
a) B cells
b) T cells
c) both a and b
d) IgA
SCID can occur due to the absence of an enzyme
a) adenosine deaminase
b) guanosine deaminase
c) phosphorylase
d) thymidine deaminase
(c)
(a)

All of the following are immunodeficiency diseases except
a) Graves disease
b) SCID
c) DiGeorge’s syndrome
d) Hyper IgE syndrome
Some defects or mutations in components of innate or adaptive immunity may lead to
a) hypersensitivity
b) auto-immune diseases
c) immunodeficiency
d) tolerance
(c)
(a)

References
Burkit’s Oral Medicine 11th edition
Textbook of oral medicine,2nd edition,Anil Ghom
A textbook of microbiology p. Chakraborty
Understanding medical physiology RL Bijlani 4th edition
McGraw-Hill’s access medicine
Essentials of Clinical Immunology, Sixth Edition. Helen Chapel, Mansel Haeney, Siraj Misbah, and Neil Snowden.© 2014 John Wiley & Sons, Ltd. Published 2014 by John Wiley & Sons, Ltd.