4 corresponding authors: 5 iris fischer (irisfischer402 ... iris fischer ([email protected])...
TRANSCRIPT

1
Running title: 1
Evolutionary dynamics of the LRR-RLK gene family 2
3
Corresponding Authors: 4
Iris Fischer ([email protected]) and Nathalie Chantret ([email protected]) 5
INRA SupAgro, UMR AGAP, 2 Place Pierre Viala, Bât. 21, 34060 Montpellier, France 6
Telephone: +33 (0)4 99 61 60 83 7
8
Research Area: Genes, Development and Evolution 9
Plant Physiology Preview. Published on January 15, 2016, as DOI:10.1104/pp.15.01470
Copyright 2016 by the American Society of Plant Biologists
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

2
Evolutionary dynamics of the Leucine-Rich Repeats Receptor-Like Kinase 10
(LRR-RLK) subfamily in angiosperms. 11
12
13
Iris Fischer1,*, Anne Diévart2, Gaetan Droc2, Jean-François Dufayard2 and Nathalie Chantret1,* 14 15 1 INRA, UMR AGAP, F-34060 Montpellier, France 16 2 CIRAD, UMR AGAP, F-34398 Montpellier, France 17 *Corresponding Authors 18
19
Summary: Phylogenetic analysis of leucine-rich repeat containing receptor-like kinases 20
demonstrates the dynamic nature of gene duplication, loss and selection in this family. 21
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

3
I.F., A.D., and N.C. designed the study; G.D. performed the LRR-RLK extraction; J-F.D. 22
performed the phylogenetic clustering; I.F., A.D., and N.C. performed the data analysis and 23
statistics; I.F. drafted the manuscript with the help of A.D. and N.C. 24
25
Financial sources: IF was funded by the German Research Foundation (DFG): FI 1984/1-1 26
and the ARCAD project (Agropolis Resource Center for Crop Conservation, Adaptation and 27
Diversity) of the Agropolis Foundation. This work was partially funded by grant #ANR-08-28
GENM-021 from Agence Nationale de la recherche (ANR, France). 29
30
Corresponding Authors: 31
Iris Fischer ([email protected]) and Nathalie Chantret ([email protected]) 32
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

4
ABSTRACT 33
Gene duplications are an important factor in plant evolution and lineage specific expanded 34
(LSE) genes are of particular interest. Receptor-like kinases (RLK) expanded massively in 35
land plants and Leucine-Rich Repeat (LRR)-RLKs constitute the largest RLK family. Based 36
on the phylogeny of 7,554 LRR-RLK genes from 31 fully sequenced flowering plant 37
genomes, the complex evolutionary dynamics of this family was characterized in depth. We 38
studied the involvement of selection during the expansion of this family among angiosperms. 39
LRR-RLK subgroups harbor extremely contrasted rates of duplication, retention or loss and 40
LSE copies are predominantly found in subgroups involved in environmental interactions. 41
Expansion rates also differ significantly depending on the time when rounds of expansion or 42
loss occurred on the angiosperm phylogenetic tree. Finally, using a dN/dS-based test in a 43
phylogenetic framework, we searched for selection footprints on LSE and single-copy LRR-44
RLK genes. Selective constraint appeared to be globally relaxed at LSE genes and codons 45
under positive selection were detected in 50% of them. Moreover, the LRR domains – and 46
specifically four amino acids in them – were found to be the main targets of positive selection. 47
Here, we provide an extensive overview of the expansion and evolution of this very large 48
gene family. 49
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

5
INTRODUCTION 50
Receptor-like kinases (RLK) comprise one of the largest gene families in plants and expanded 51
massively in land plants (Embryophyta) (Lehti-Shiu et al., 2009; Lehti-Shiu et al., 2012). For 52
plant RLK gene families, the functions of most members are often not known (especially in 53
recently expanded families) but some described functions include innate immunity (Albert et 54
al., 2010), pathogen response (Dodds and Rathjen, 2010), abiotic stress (Yang et al., 2010), 55
development (De Smet et al., 2009), and sometimes multiple functions (Lehti-Shiu et al., 56
2012). The RLKs usually consist of three domains: an amino-terminal extracellular domain 57
(ECD), a transmembrane (TM) domain, and a carboxy-terminal kinase domain (KD). In 58
plants, the KD usually has a serine/threonine specificity (Shiu and Bleecker, 2001) but 59
tyrosine specific RLKs were also described (e.g. BRASSINOSTEROID INSENSITIVE1 (Oh 60
et al., 2009)). Interestingly, it was estimated that ~20% of RLKs contain a catalytically 61
inactive KD (e.g. STRUBBELIG, CORYNE (Chevalier et al., 2005; Castells and 62
Casacuberta, 2007; Gish and Clark, 2011)). In Arabidopsis, 44 RLK subgroups (SG) were 63
defined by inferring the phylogenetic relationships between the KDs (Shiu and Bleecker, 64
2001). Interestingly, different SGs show different duplication/retention rates (Lehti-Shiu et 65
al., 2009). Specifically, RLKs involved in stress response show a high number of tandemly 66
duplicated genes whereas those involved in development do not (Shiu et al., 2004) which 67
suggests that some RLK genes are important for responses of land plants to a changing 68
environment (Lehti-Shiu et al., 2012). There seem to be relatively few RLK pseudogenes 69
compared to other large gene families and copy retention was argued to be driven by both, 70
drift and selection (Zou et al., 2009; Lehti-Shiu et al., 2012). As most SGs are relatively old 71
and RLK subfamilies expanded independently in several plant lineages, duplicate retention 72
cannot be explained by drift alone and natural selection is expected to be an important driving 73
factor in RLK gene family retention (Lehti-Shiu et al., 2009). 74
Leucine-Rich Repeat (LRR)-RLKs, which contain up to 30 LRRs in their extracellular 75
domain, constitute the largest RLK family (Shiu and Bleecker, 2001). Based on the kinase 76
domain, 15 LRR-RLK SGs have been established in Arabidopsis (Shiu et al., 2004; Lehti-77
Shiu et al., 2009). So far, two major functions have been attributed to them: defense against 78
pathogens and development (Tang et al., 2010b). LRR-RLKs involved in defense are 79
predominantly found in lineage specific expanded (LSE) gene clusters whereas LRR-RLKs 80
involved in development are mostly found in non-expanded groups (Tang et al., 2010b). It 81
was also discovered that the LRR domains are significantly less conserved than the remaining 82
domains of the LRR-RLK genes (Tang et al., 2010b). In addition, a study on four plant 83
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

6
genomes (A. thaliana, grape, poplar, rice) showed that LRR-RLK genes from LSE gene 84
clusters show significantly more indication of positive selection or relaxed constraint than 85
LRR-RLKs from non-expanded groups (Tang et al., 2010b). 86
The genomes of flowering plants (angiosperms) have been shown to be highly dynamic 87
compared to most other groups of land plants (Leitch and Leitch, 2012). This dynamic is 88
mostly caused by the frequent multiplication of genetic material, followed by a complex 89
pattern of differential losses (i.e. the fragmentation process) and chromosomal rearrangements 90
(Langham et al., 2004; Leitch and Leitch, 2012). Most angiosperm genomes sequenced so far 91
show evidence for at least one whole genome multiplication event during their evolution (see 92
e.g. Jaillon et al. (2007); D'Hont et al. (2012); The Tomato Genome Consortium (2012)). At a 93
smaller scale, tandem and segmental duplications are also very common in angiosperms (The 94
Arabidopsis Genome Initiative, 2000; International Rice Genome Sequencing Project, 2005; 95
Rizzon et al., 2006). Although the most common fate of duplicated genes is to be 96
progressively lost, in some cases they can be retained in the genome and adaptive as well as 97
non-adaptive scenarios have been discussed to play a role in this preservation process (for 98
review see Moore and Purugganan (2005); Hahn (2009); Innan (2009); Innan and Kondrashov 99
(2010)). Whole genome sequences also revealed that the same gene may undergo several 100
rounds of duplication and retention. These lineage specific expanded genes were shown to 101
evolve under positive selection more frequently than single-copy genes in angiosperms 102
(Fischer et al., 2014). The study by Fischer et al. analyzed general trends over whole 103
genomes. Here, we ask if, and to what extent, this trend is observable at LRR-RLK genes. As 104
this gene family is very dynamic and large – and in accordance with the results of Tang et al. 105
(2010b) – we expect the effect of positive selection to be even more pronounced than in the 106
whole genome average. 107
We analyzed 33 Embryophyta genomes to investigate the evolutionary history of the 108
LRR-RLK gene family in a phylogenetic framework. Twenty LRR-RLK subgroups were 109
identified and from this dataset we deciphered the evolutionary dynamics of this family within 110
angiosperms. The expansion/reduction rates were contrasted between SGs and species as well 111
as in ancestral branches of the angiosperm phylogeny. We then focused on genes which 112
number increased dramatically in a subgroup- and/or species-specific manner (i.e. LSE 113
genes). Those genes are likely to be involved in species-specific cellular processes or adaptive 114
interactions and were used as a template to infer the potential occurrence of positive selection. 115
This led to the identification of sites on which positive selection likely acted. We discuss our 116
results in the light of angiosperm genome evolution and current knowledge of LRR-RLK 117
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

7
functions. Positive selection footprints identified in LSE genes highlight the importance of 118
combining evolutionary analysis and functional knowledge to guide further investigations. 119
120
121
122
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

8
RESULTS 123
We extracted genes containing both LRRs and a KD from 33 published Embryophyte 124
genomes. Here, we mostly describe the findings for the 31 angiosperm (eight monocot and 23 125
dicot) genomes we analyzed. The 7,554 LRR-RLK genes were classified in 20 subgroups 126
(SGs). This classification was inferred using distance related methods because the high 127
number of sequences to be analyzed would imply excessive computation time for methods 128
relying on maximum likelihood. Since we decided to study the evolutionary dynamic of LRR-129
RLK gene family using the SG classification as a starting point, we first wanted to verify that 130
each SG was monophyletic. Ten subsets of about 750 sequences were created by picking one 131
sequence out of ten to infer a PHYML tree (data not shown). Analysis of the trees shows that 132
most SGs (14) are monophyletic with strong branch support. On the other hand, for six SGs 133
(SG_I, SG_III, SG_VI, SG_Xb, SG_XI and SG_XV), the topology differs slightly between 134
trees: in at least five trees out of ten, either the SG appears to be paraphyletic or few 135
sequences are placed outside the main monophyletic clade with low branch support. As we 136
could not confirm that these SG are monophyletic, they were tagged with a "*" throughout the 137
manuscript. 138
Next, we determined the number of ancestral genes present in the last common ancestor 139
of angiosperms (LCAA) using a tree reconciliation approach (see Materials and Methods). In 140
short, tree reconciliation compares each SG-specific LRR-RLK gene tree to the species tree to 141
infer gene duplications and losses. Note that since only LRR-RLKs with at least one complete 142
LRR were considered, some of the inferred gene losses might correspond to RLKs without, or 143
with degenerated LRRs. Using this method, we predicted the number of LRR-RLK genes in 144
the LCAA to be 150. All SGs were present in the LCAA but the number of genes between 145
SGs was highly variable (Table I). SG_III* and SG_XI* show the highest number of ancestral 146
genes, with 32 and 29 genes, respectively. The lowest numbers of ancestral genes are 147
recorded for SG_VIIb, SG_Xa, SG_XIIIa, and SG_XIIIb which only possessed two genes and 148
SG_XIV which only contained one. These results show that already in the LCAA that lived 149
~150 million years ago (Supplemental Table SI) some SGs were more prone to retain copies 150
than others. We wanted to determine if this ancestral pattern was preserved during the course 151
of angiosperm evolution and if different SGs expanded or contracted compared to the LCAA. 152
153
Expansion rates of LRR-RLK genes differ between subgroups and species 154
To gain a more comprehensive understanding of LRR-RLK evolution, we first looked at SG-155
specific expansion rates in two complementary ways. First, we calculated the global SG 156
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

9
expansion rate (= ratio of contemporary LRR-RLK genes per species in one SG divided by 157
the ancestral number) for each SG (Fig. 1). Second, we inferred the branch-specific expansion 158
rate of each SG on the phylogenetic tree of the 31 angiosperm species. We did this by 159
automatically computing the ratio of descendant LRR-RLKs divided by the ancestral number 160
of LRR-RLKs at every node (see Materials and Methods) (Fig. 2). Looking at the global SG 161
expansion, we found that SG_Xa, SG_XIIa, SG_XIIb, and SG_XIV expanded more than 2-162
fold on average, and SG_I* and SG_IX around 2-fold (Fig. 1, Supplemental Table SII). 163
Interestingly, SG_XIIa already had a moderate-high ancestral gene number (nine) and 164
therefore seems to be generally prone to high retention rates. Indeed, SG_XIIa was subject to 165
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

10
repeated rounds of major expansion events (i.e. expansion > 2-fold) during its evolutionary 166
history, e.g. in Poaceae, the Solanum ancestor, Malvaceae, and the Arabidopsis ancestor; but 167
also species-specific expansions, e.g. in THECC, GOSRA, ARALY, SCHPA, MALDO, 168
LOTJA, POPTR, and JATCU (Fig. 2, see Table II for five-digit species code). On the other 169
hand, SG_I* and SG_XIIb had a medium number of copies in the ancestral genome (seven 170
and four, respectively) but the pattern of expansion is quite different when analyzed in detail 171
(Fig. 2). For SG_I*, the expansion rate is mostly due to ancestral expansion events rather than 172
species-specific ones. For example, the high number of copies in ARATH and EUTSA (Fig. 173
1) is not due to expansions specific to these species but rather an expansion in Brassicaceae. 174
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

11
Subsequently, copies were lost in the other species of this family analyzed here (ARALY, 175
SCHPA, BRARA) but retained in ARATH and EUTSA (Fig. 2). Species-specific expansions 176
can also be observed in SG_I*, mostly in PRUPE and POPTR. For SG_XIIb, on the other 177
hand, the high expansion rate is mostly due to recent species-specific expansions in PHODC, 178
MUSAC, VITVI, GOSRA, MALDO, POPTR, and JATCU. But one major ancestral 179
expansion can be observed in Rosids. 180
SG_IX, SG_Xa, and SG_XIV had only few copies in the LCAA (three, two, and one, 181
respectively) and all show a relatively high global expansion rate (Fig. 1). For these 182
subgroups also, a contrasted branch-specific expansion patterns can be observed (Fig. 2). 183
SG_Xa went through relatively few major expansions: one can be detected in the dicots 184
ancestor and a species-specific one in POPTR. Likewise, SG_IX shows only one ancestral 185
expansion in Malvaceae but more species-specific expansions in PHODC, MUSAC, 186
MALDO, and GLYMA. Finally, SG_XIV went through several rounds of ancestral 187
(monocots, dicots, Malvaceae, and Brassicaceae) as well as species-specific expansion 188
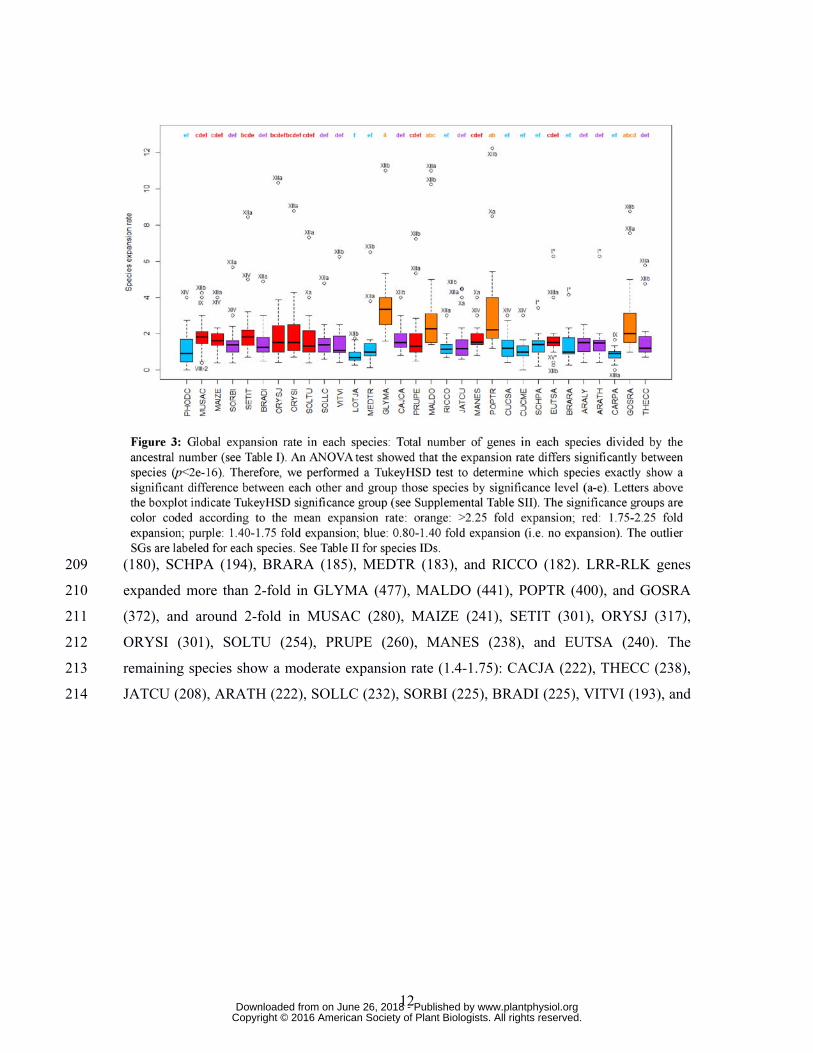
(PHODC, MALDO, and POPTR). The other SGs show a moderate expansion rate (1.3-1.75) 189
or no expansion at all (Fig. 1, Supplemental Table SII). SG_XV* is the only SG for which the 190
number of copies was decreasing on average compared to the LCAA genome (0.77). It is 191
important to note that the LCAA ancestral gene number could have been slightly over-192
estimated for those SGs without a confirmed monophyletic origin (denoted by “*”), resulting 193
in an under-estimation of global expansion rate. However, we re-calculated the global 194
expansion rates for each of those SGs using the largest subset of sequence that always include 195
a stable monophyletic clade. The obtained global expansion rate differed only slightly from 196
the ones presented here (data not shown) and the conclusions drawn remain unchanged. 197
Because some species underwent whole genome duplication (WGD) or whole genome 198
triplication (WGT) relatively recently compared to others (Table III), we determined species-199
specific patterns of LRR-RLK expansions and looked if those patterns are consistent with the 200
recent history of the species. Therefore, we computed the global species expansion rate (= 201
ratio of LRR-RLK genes per SG in one species divided by the ancestral number) for each of 202
the 31 angiosperm species. As expected, the global expansion rate differs significantly 203
between species (Fig. 3, Supplemental Table SII). Compared to the LCAA (150 genes), the 204
number of LRR-RLK genes did not decrease for most species except for LOTJA (114) and 205
CARPA (127). This indicates that, on average, LRR-RLK genes are more prone to retention 206
than loss. Some species, however, did not significantly expand their average number of LRR-207
RLK genes compared to the common ancestor: PHODC (158), CUCME (149), CUCSA 208
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.
12
(180), SCHPA (194), BRARA (185), MEDTR (183), and RICCO (182). LRR-RLK genes 209
expanded more than 2-fold in GLYMA (477), MALDO (441), POPTR (400), and GOSRA 210
(372), and around 2-fold in MUSAC (280), MAIZE (241), SETIT (301), ORYSJ (317), 211
ORYSI (301), SOLTU (254), PRUPE (260), MANES (238), and EUTSA (240). The 212
remaining species show a moderate expansion rate (1.4-1.75): CACJA (222), THECC (238), 213
JATCU (208), ARATH (222), SOLLC (232), SORBI (225), BRADI (225), VITVI (193), and 214
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

13
ARALY (195). As expected, the four species with the highest global expansion rate 215
(GLYMA, MALDO, POPTR, GOSRA) are recent polyploids in which most SGs have 216
expanded (Fig. 2). However, some SGs expanded more than 2-fold, indicating that small scale 217
duplication events have occurred in addition to polyploidy. In POPTR, for instance, the global 218
expansion rates of SG_Xa and SG_XIIb are more than 8-fold (Fig. 3) and a strong branch-219
specific expansion rate is detected on the terminal POPTR branch (3.25 for SG_Xa and 5.4 220
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

14
for SG_XIIb) (Fig. 2). Surprisingly, SG_VIIa and SG_VIIb show a high branch-specific 221
expansion rate in POPTR (4.0 and 3.0, respectively), which is not reflected in the global 222
expansion rate in this species (Fig. 3). This is due to the fact that SG_VIIa and SG_VIIb went 223
through strong reduction in Malpighiales (0.33) and Fabids (0.5), respectively. Thus, the 224
cumulative effect of successive reductions and expansions is not evident in the global 225
expansion rate. These contrasted evolutionary dynamics can also be observed in MALDO. A 226
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

15
global expansion of SG_IX was not detected because of the strong reduction in 227
Amygdaloideae. To summarize, this data can be integrated into the species phylogeny to draw 228
an image of the complex evolutionary dynamics of the LRR-RLK gene family through time 229
(Fig. 4). 230
231
Different patterns of lineage specific expansion in LRR-RLK subgroups 232
Given the differences of LRR-RLK expansion rates between species, we wanted to identify 233
cases of LSE, i.e. cases where a high duplication/retention rate is specific to one species. 234
Using a tree reconciliation approach (see Materials and Methods), we built a dataset 235
consisting of ultraparalog clusters (UP – only related by duplication) which represents the 236
LSE events and a superortholog reference gene set (SO – only related by speciation). We only 237
considered clusters containing five or more sequences. After cleaning, our final dataset 238
comprised 75 UP and 189 SO clusters containing 796 and 1,970 sequences, respectively 239
(Table IV). The median number of sequences in the UP clusters is not significantly different 240
from the median number in SO clusters (8 in both cases) (Supplemental Fig. S1). For UP 241
clusters, however, the alignments are significantly longer (Mann-Whitney test: p<0.001) with 242
a median of 3,237 base pairs (bp) compared to 2,841 for SO clusters. One possible 243
explanation for this could be that UP clusters are more dynamic and might contain more 244
LRRs. PRANK, the alignment algorithm we used, introduces gaps instead of aligning 245
ambiguous sites and therefore produces longer alignments when sequences are divergent. 246
However, this phenomenon does not influence the outcome of further test for positive 247
selection using codeml (Yang, 2007). 248
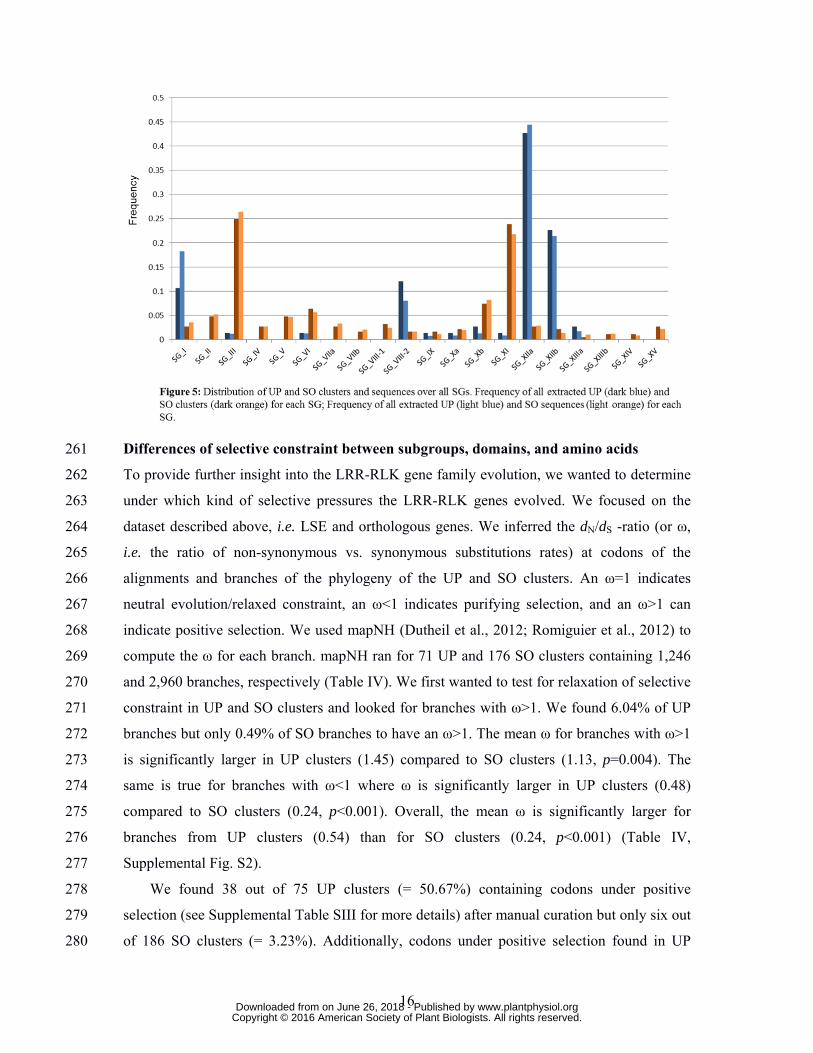
We then wanted to determine which SGs are represented in the SO and UP dataset. 249
Unsurprisingly, all SGs were present in SO clusters (Fig. 5). This could be expected as all 250
SGs were already present in the LCAA and remained stable or expanded (except SG_XV*). 251
In general, the frequency of SO clusters (and sequences) for each SG reflects the number of 252
copies in the LCAA (Table I, Fig. 5). On the other hand, only eleven of the 20 subgroups 253
were represented in UP clusters (SG_I*, SG_III*, SG_VI*, SG_VIII-2, SG_IX, SG_Xa, 254
SG_Xb*, SG_XI*, SG_XIIa, SG_XIIb, SG_XIIIa) and these SGs harbor a total of 837 255
sequences. SG_I*, SG_VIII-2, SG_XIIa, and SG_XIIb are clearly over-represented which is 256
in accordance with their expansion pattern. Other expanded SGs, however, have only a low 257
number of UP clusters or – in the case of SG_IV – no UP clusters at all. Therefore, it seems 258
that recently duplicated genes are more prone to be retained in some SGs. 259
260
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.
16
Differences of selective constraint between subgroups, domains, and amino acids 261
To provide further insight into the LRR-RLK gene family evolution, we wanted to determine 262
under which kind of selective pressures the LRR-RLK genes evolved. We focused on the 263
dataset described above, i.e. LSE and orthologous genes. We inferred the dN/dS -ratio (or ω, 264
i.e. the ratio of non-synonymous vs. synonymous substitutions rates) at codons of the 265
alignments and branches of the phylogeny of the UP and SO clusters. An ω=1 indicates 266
neutral evolution/relaxed constraint, an ω<1 indicates purifying selection, and an ω>1 can 267
indicate positive selection. We used mapNH (Dutheil et al., 2012; Romiguier et al., 2012) to 268
compute the ω for each branch. mapNH ran for 71 UP and 176 SO clusters containing 1,246 269
and 2,960 branches, respectively (Table IV). We first wanted to test for relaxation of selective 270
constraint in UP and SO clusters and looked for branches with ω>1. We found 6.04% of UP 271
branches but only 0.49% of SO branches to have an ω>1. The mean ω for branches with ω>1 272
is significantly larger in UP clusters (1.45) compared to SO clusters (1.13, p=0.004). The 273
same is true for branches with ω<1 where ω is significantly larger in UP clusters (0.48) 274
compared to SO clusters (0.24, p<0.001). Overall, the mean ω is significantly larger for 275
branches from UP clusters (0.54) than for SO clusters (0.24, p<0.001) (Table IV, 276
Supplemental Fig. S2). 277
We found 38 out of 75 UP clusters (= 50.67%) containing codons under positive 278
selection (see Supplemental Table SIII for more details) after manual curation but only six out 279
of 186 SO clusters (= 3.23%). Additionally, codons under positive selection found in UP 280
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

17
clusters are not distributed evenly over domains (Fig. 6). To account for differences in domain 281
size, a hit frequency, i.e. the number of sites under positive selection we found relative to all 282
sites possible for each domain, was calculated (see Materials and Methods). The domain 283
showing the highest hit frequency is the LRR domain, followed by the cysteine-pairs and their 284
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

18
flanking regions (Fig. 6A). Hits in both domains are distributed over all SGs and species 285
tested. The KD and its surrounding domains contain very few codons under positive selection. 286
Domains classified as “other” combine domains important for the function of the LRR-RLK 287
genes but vary between SGs. For example, SG_I* (Fig. 6B) contains a malectin domain. All 288
hits classified as “other” here fall in the malectin-like domain (MLD) of a POPTR SG_I* 289
cluster. 290
Finally, we wanted to investigate whether some AAs in the LRR are more frequently 291
targeted by positive selection. The LRR typically contains 24 AAs and sometimes islands 292
between them (Fig. 6C). Four AAs were predominantly subject to positive selection: 6, 8, 10, 293
and 11 which all lie in the LRR-characteristic LXXLXLXX β-sheet/β-turn structure. 294
295
296
297
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

19
DISCUSSION 298
We studied the SG- and species-specific expansion dynamics in LRR-RLK genes from 31 299
angiosperm genomes in a phylogenetic framework. We also analyzed the lineage-specifically 300
expanded genes in this family to determine to which extent positive selection occurred on 301
them using a dN/dS-based test. We found differences in expansion patterns depending on SGs 302
and species but only a few SGs that were subjected to LSE. A significantly higher proportion 303
of LSE LRR-RLK genes was affected by positive selection compared to single-copy genes 304
and the LRR domain (specifically four AAs within this domain) were targeted by positive 305
selection. In the following, we will discuss our findings in more detail. 306
307
Subgroup- and species-specific expansions 308
We observed significant variations in the global expansion rates between LRR-RLK SGs. 309
These are due to a complex history of expansion-retention-loss cycles that are specific to each 310
SG. The phylogenetic approach allowed us to determine the relative importance of ancestral 311
versus recent species-specific expansions for each SG and to characterize precisely the 312
loss/retention dynamics along evolutionary history of the studied species (summarized in Fig. 313
4). For example, SG_III* and SG_XI* had a high copy number of LRR-RLK in the LCAA 314
and kept a stable copy number over the last 150 million years. On the other hand, SG_I*, 315
SG_XIIa, and SGXIIb, which had a moderate copy number in the LCAA, keep expanding. 316
Some functions have been described for genes of these SGs, mainly in A.thaliana 317
(Supplemental Table SIV). For SG_III* and SG_XI*, mostly genes involved in development 318
are described. The high numbers of ancestral genes in these two SGs combined with their size 319
stability during angiosperm evolution may be interpreted as an early high level of 320
diversification/specialization of these genes which are needed to orchestrate common 321
developmental features. This hypothesis can be reinforced by the high number of 322
superorthologous genes in these subgroups. For SG_I* and SG_XIIa, on the other hand, 323
mostly genes involved in response to biotic stress are described up to now. These observations 324
confirm that different expansion/retention patterns appear to be related to gene function 325
although one has to keep in mind that functions have only been assigned to few LRR-RLK 326
genes. Three SGs (SG_IX, SG_Xa, and SG_XIV) expanded compared to their very low 327
ancestral number (1-3) leading to a high total expansion rate. As it has been postulated that 328
duplications are the raw material for adaptation (Nei and Rooney, 2005; Fischer et al., 2014), 329
the evolution of those SGs was likely driven by adaptation – to varying degrees in different 330
angiosperm species, depending on the environment they evolved in. The known functions are 331
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

20
both related to response to biotic or abiotic stress and development. Because so far our 332
knowledge of LRR-RLK functions is limited and mostly restricted to A. thaliana, further 333
studies are needed to make more reliable statements on the link between function and 334
expansion/retention dynamics in different SGs. 335
Next, we wanted to ascertain species-specific expansions of LRR-RLK genes and how 336
they are related to the recent history of the species in our study. Whole genome multiplication 337
has been argued to be a major force in diversification of angiosperms (Soltis et al., 2009; 338
Soltis and Burleigh, 2009; Renny-Byfield and Wendel, 2014). All angiosperms share two 339
ancient WGDs (Jiao et al., 2011). Likewise, all monocots share a WGD ~130 Mya (Tang et 340
al., 2010a) and most dicots (Eudicots) share a WGT around the same time (Jaillon et al., 341
2007; Wang et al., 2012), but more recent WGDs and WGTs occurred in many angiosperm 342
species (Fig. 4, Table III). The link between WGD/Ts and the number of LRR-RLK genes is 343
not straightforward. We found that in Glycine max, Gossypium raimondii, and Malus x 344
domestica, which were subject to relatively recent WGDs (15-13, 17-13 and 45-30 Mya, 345
respectively) (Pfeil et al., 2005; Velasco et al., 2010; Wang et al., 2012), the number of LRR-346
RLK genes expanded more than 2-fold compared to the LCAA. These results are in 347
accordance with what was already described for these species. Indeed, it was found that G. 348
max contains a very large number of retained genes from this WGD (Cannon et al., 2014). 349
Additionally, recent studies on large gene families in G. raimondii indicate that their copy 350
number is either driven by retention after the last WGD (e.g. NAC transcription factors) 351
(Shang et al., 2013) or a combination of segmental (SD) and tandem duplications (TD) (e.g. 352
WRKY transcription factors) (Dou et al., 2014). For M. domestica (most recent WDG after 353
the divergence for peach according to Verde et al. (2013)), a recent study on nucleotide 354
binding site (NBS) LRR genes showed that they also stem mostly from SDs and TDs (Arya et 355
al., 2014). 356
More contrasted results are observed in Brassicaceae where two WGDs occurred (Barker 357
et al., 2009; Fawcett et al., 2009). Most SGs expand their number of genes on this ancestral 358
branch, but the species belonging to this clade mostly retain or loose genes on average (Fig. 2 359
and 4). The only exception concerns Eutrema salsugineum (an A. thaliana relative) which is 360
the only species with a more than 2-fold average expansion rate. The global expansion rate in 361
E. salsugineum is mostly due to two SGs (SG_I* and SG_XIIIa). In the original genome 362
paper (Wu et al., 2012), the authors found that genes from the category “response to stimulus” 363
(response to salt stress, osmotic stress, water deprivation, ABA stimulus, and hypoxia) are 364
significantly over-represented in E. salsugineum compared to A. thaliana. This over-365
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

21
representation is described as mostly caused by SDs and TDs (Wu et al., 2012) in accordance 366
with what we observed in SG_XIIIa. This could be of functional importance to this halophile 367
plant. 368
Finally, of all species analyzed here, Zea mays and Brassica rapa (and maybe Manihot 369
esculenta) show the most recent cases of WGD/T (12-5 and 9-5 Mya, respectively) (Schnable 370
et al., 2011; Wang et al., 2011) yet their expansion rates are moderate. This is further evidence 371
for the dynamic nature of angiosperm genomes that has been discussed before (Leitch and 372
Leitch, 2012; Fischer et al., 2014). After a WGD event, genomes tend to return to the diploid 373
(or previous) state by losing redundant duplicated genes (fractionation process) – although the 374
gene loss is biased (Bowers et al., 2003; Schnable et al., 2009). Which genes are lost or 375
retained strongly depends on their function (De Smet et al., 2013). However, it has been 376
shown that genes involved in stress response are mostly created by TDs rather than WGD 377
(Hanada et al., 2008). Indeed, it was hypothesized before that RLK genes involved in stress 378
response mostly duplicate by TD (Shiu et al., 2004). Here, we provide a detailed 379
representation of expansion-retention-loss dynamics of the whole LRR-RLK gene family in 380
31 angiosperm species (Fig.4). Each new genome sequenced will improve the accuracy of the 381
expansion-retention-loss event predictions and will help identifying new elements that can be 382
useful for future functional analysis and/or linked to adaptive traits. 383
384
Studying selection pressures in a large and dynamic gene family 385
As described above, the composition of LRR-RLKs in each of the 31 studied angiosperm 386
species results from a complex dynamic of species- and SG-specific expansion/loss events. To 387
further investigate the potential role of this family in plant adaptation we analyzed to which 388
selective pressures the LRR-RLKs were submitted. Such an analysis cannot be considered for 389
the phylogeny of the entire gene family because of the high number of sequences and high 390
sequence divergence (the phylogeny on which we divided the SGs was inferred on the 391
conserved kinase domain only). We then chose to focus on two specific cases: (i) LSE as a 392
specific case of duplication/retention, and (ii) a subset of strictly orthologous genes. Indeed, 393
LSE has been shown to fuel adaptation in angiosperms (Fischer et al., 2014) and we wanted to 394
test the prevalence of this mode of duplication in our large dataset. Therefore, we evaluated to 395
which extant LRR-RLK genes were subject to LSE and how positive selection acted on those 396
genes. As a reference, we chose the strictly orthologous subset. This approach allows the 397
interpretation of LSE evolution compared to the general LRR-RLK selective background 398
(Fischer et al., 2014). 399
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

22
The power of this phylogenetic approach relies on the number of species analyzed and we 400
profit from an ever increasing number of sequenced plant genomes. Another important 401
requirement for this approach is the quality of sequencing and annotation – especially for a 402
large gene family – as sequencing errors and mis-annotations can lead to false positives when 403
testing for positive selection (Han et al., 2013). We profit from a recently developed pipeline 404
designed to automatically perform different steps of the analysis (Fischer et al., 2014). This 405
allowed us to quickly incorporate sequenced genomes of choice and future studies can easily 406
expand this analysis as new reliable data becomes available. Finally, we set great value on 407
manually verifying the data throughout the process – from the identification of the LRR-408
RLKs to the inference of positive selection. Although this is tedious work for such a large 409
dataset, it is nevertheless important. As we recently showed, ~50% automatically reported 410
instances of positive selection turned out to be false positives after manual curation (Fischer et 411
al., 2014). 412
We found that all SGs are represented in the single-copy reference set with an over-413
representation of SG_III* and SG_XI*. This is in accordance with the fact that these two SGs 414
had the highest number of copies in the genome of the LCAA and did not significantly 415
expand since (see above). In general, the frequency of clusters from the single-copy gene set 416
(and sequences) for each SG reflects the number of copies in the LCAA (Table I, Fig. 5). On 417
the other hand, only eleven of the 20 SGs were represented in the LSE dataset. This is mainly 418
because the majority of expansions are rather old in these SGs, whereas they happened 419
relatively recently in SG_I*, SG_VIII-2, SG_XIIa, and SG_XIIb (see above). Fourteen 420
species (or clades) are represented in the LSE dataset: MUSAC (2 ultraparalog clusters), 421
SETIT (1), ORYZA (10), VITVI (3), SOLAN (6), MEDTR (3), GLYMA (2), PRUPE (6), 422
MALDO (11), POPTR (8), BRASS (11), GOSRA (5), THECC (2), and PHYPA (5). Again, 423
not every species is affected to the same extent, but this does not necessarily reflect recent 424
WGD/T. Additionally, LSE can also arise from SD and TD which frequency of occurrence is 425
not uniform within or between genomes. Our results indicate that different species are more 426
prone to retain recently duplicated genes than others. This in turn might reflect on their recent 427
evolution or domestication which should be examined in more detail in future studies. 428
When focusing on the study of selective pressures, we first looked at ω at the branches of 429
the LSE and single-copy gene clusters and found that selective constraint was relaxed in the 430
LSE dataset. This outcome was expected as it was previously shown that LSE genes evolve 431
more relaxed constraint than single-copy genes in angiosperms (Fischer et al., 2014). This 432
study, however, looked at whole angiosperm genomes but a similar pattern has already been 433
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

23
demonstrated in other large gene families (e.g. Johnson and Thomas, 2007; Xue et al., 2012; 434
Yang et al., 2013a; Yang et al., 2013b) and in LRR-RLK genes in particular (Tang et al., 435
2010b). Previous studies on that subject only had a limited dataset (four angiosperm species; 436
Tang et al. 2010b). Here, we demonstrate that this is still true when a larger and more 437
representative sample of angiosperms is considered. 438
Next, we wanted to identify codons which evolved under positive selection in the LSE 439
and the single-copy dataset. A recent study on gene families in the whole genomes of ten 440
angiosperms found that 5.4% of LSE genes contained codons showing positive selection 441
footprints (Fischer et al., 2014). Here, we ask if and to what extend this is also true for the 442
large and dynamic LRR-RLK gene family. We discovered that for LSE LRR-RLK genes, the 443
rate of codons under selection is almost 10-fold higher (50.67%) than the genome average. In 444
addition, we found >3% of single copy genes containing codons under selection whereas 445
Fischer et al. (2014) described no case of positive selection at the single-copy gene clusters in 446
their study. Together with the high rate of branches with ω>1 in LSE gene clusters (6.04%, 447
compared to 0.49% for single-copy genes) this indicates that LRR-RLK genes are more prone 448
to evolve under positive selection than the average of angiosperm gene families. As it might 449
be expected, all ultraparalog clusters with codons under positive selection come from the four 450
over-represented SGs: SG_I* (1 ultraparalog cluster), SG_VIII-2 (3), SG_XIIa (24), and 451
SG_XIIb (10). The single-copy gene clusters with codons under selection come from six SGs: 452
SG_III, SG_VIIa, SG_Xa, SG_Xb, SG_XIIa, and SG_XIIb. Therefore, recent expansion and 453
retention only affect a few SGs but in those SGs positive selection plays an important role. 454
For SG_XIIa, positive selection has been already previously inferred for genes involved in 455
environmental interactions: Xa21, which confers resistance to the bacterial blight disease, was 456
found to have evolved under positive selection in rice (Wang et al., 1998; Tan et al., 2011); 457
and FLS2, involved also in response to biotic stress, shows a signature of rapid fixation of an 458
adaptive allele in Arabidopsis (Vetter et al., 2012). Future studies on smaller subsets of SGs 459
will surely cast further light on selection patterns in LRR-RLK genes. Only eleven species (or 460
clades) are represented in the LSE dataset with codons under positive selection: SETIT (1 461
ultraparalog cluster), ORYZA (2), SOLAN (4), MEDTR (2), GLYMA (2), PRUPE (3), 462
MALDO (8), POPTR (7), BRASS (2), GOSRA (5), and THECC (2). Not every species is 463
affected the same level by positive selection and again future studies might bring more details 464
concerning the evolutionary history of specific species and SGs to light. 465
In addition, we found that not every domain of the LRR-RLK genes was similarly 466
affected by positive selection. Most codons under selection fall in the LRR domain. This 467
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

24
outcome might be expected as LRRs are very dynamic and plasticity in this region provides 468
plants with a broad toolset to face environmental challenges and therefore undergoes positive 469
selection frequently (Zhang et al., 2006; Tang et al., 2010b). Only very few codons under 470
positive selection were found in the kinase domain and its surrounding regions. This result is 471
consistent with the fact that the KD is very conserved among species and SGs and evolved 472
mostly under purifying selection (Shiu et al., 2004; Tang et al., 2010b). A more surprising 473
result was the identification of a significant number of positively selected sites in the 474
malectin-like domain of a Populus trichocarpa SG_I* cluster. So far, the function of 475
extracellular malectin-like domains of RLKs is not well understood (Lindner et al., 2012). 476
However, a malectin-like domain-containing SG_I* LRR-RLK has been described to confer 477
susceptibility to a downy mildew pathogen in A. thaliana and to have similarities to 478
Symbiosis RLKs (SYMRKs) which are important for the regulation of bacterial symbiont 479
accommodation (Markmann et al., 2008; Hok et al., 2011). Therefore, our results suggest that 480
it could be interesting to further investigate the function and evolutionary history of this 481
SG_I* domain, particularly in P. trichocarpa. Another unexpected finding was the frequent 482
occurrence of positive selection at the cys-pairs and flanking regions which are involved in 483
folding and/or the binding to other proteins. To what extent the function of LRR-RLKs is 484
affected by mutations in the cys-pair regions depends on the function of the gene (Song et al., 485
2010; Sun et al., 2012) and it would be interesting to study this in more detail in the future. 486
Finally, we took a closer look at the AAs in the LRR primarily affected by positive 487
selection. Only four, out of the 24 AAs a LRR typically contains, were predominantly and 488
strongly subject to positive selection. These variable AAs lie in the un-conserved part of the 489
LRR-characteristic LXXLXLXX β-sheet/β-turn structure which is involved in protein-protein 490
interactions (Jones and Jones, 1997; Enkhbayar et al., 2004). Specifically, solvent-exposed 491
residues were targeted by positive selection (Parniske et al., 1997; Wang et al., 1998). Further 492
investigation on the functional consequences of these nucleotide variations need to be done to 493
confirm their adaptive potential but our findings align very well to the current understanding 494
of LRR ligand binding. Taken together, our results could be very useful for further functional 495
investigations of LRR-RLK genes in different species. 496
497
498
CONCLUSIONS 499
We studied LRR-RLK genes from 33 land plant species to investigate SG- and species-500
specific expansion of these genes, to which extent they were subject to LSE, and to determine 501
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

25
the role positive selection played in the evolution of this large gene family. We described that 502
some SGs are more prone to expansion/retention than others and that the expansions occurred 503
at different times in the evolution of LRR-RLK genes. This fine-scale analysis of the dynamic 504
allowed us to identify branches and species for which a higher than average retention rate 505
could indicate a potential adaptive event for some SGs. We also described that only a few SGs 506
show patterns of recent LSE and that at those genes selective constraint is relaxed. More than 507
50% of the LSE genes contain codons which show evidence for positive selection which is 508
almost 10-fold the frequency previously described at gene families in angiosperms (Fischer et 509
al., 2014). Finally, we found that over the LRR-RLK genes the LRR domain and specifically 510
four AAs responsible for ligand interaction are most frequently subject to selection. 511
512
513
MATERIALS AND METHODS 514
Studied Genomes 515
We analyzed 31 angiosperm genomes (eight monocot (sub)species and 23 dicot species) (see 516
Table II): Phoenix dactylifera (Al-Dous et al., 2011), Musa acuminata (D'Hont et al., 2012), 517
Oryza sativa subsp. japonica (International Rice Genome Sequencing Project, 2005), Oryza 518
sativa subsp. indica (Yu et al., 2002), Brachypodium distachyon (The International 519
Brachypodium Initiative, 2010), Zea mays (Schnable et al., 2009), Sorghum bicolor (Paterson 520
et al., 2009), Setaria italica (Zhang et al., 2012), Solanum tuberosum (Potato Genome 521
Sequencing Consortium, 2011), Solanum lycopersicum (The Tomato Genome Consortium, 522
2012), Vitis vinifera (Jaillon et al., 2007), Lotus japonicus (Sato et al., 2008), Cajanus cajan 523
(Varshney et al., 2012), Arabidopsis thaliana (The Arabidopsis Genome Initiative, 2000), 524
Arabidopsis lyrata (Hu et al., 2011), Schrenkiella parvula (a synonym is Eutrema parvula, we 525
used the nomenclature from Oh et al. (2014)) (Dassanayake et al., 2011), Eutrema 526
salsugineum (a synonym is Thellungiella halophila, we chose the nomenclature according to 527
phytosome: http://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Esalsugineum) (Wu 528
et al., 2012), Brassica rapa (Wang et al., 2011), Populus trichocarpa (Tuskan et al., 2006), 529
Glycine max (Schmutz et al., 2010), Medicago truncatula (Young et al., 2011), Prunus 530
persica (Ahmad et al., 2011), Malus x domestica (Velasco et al., 2010), Ricinus communis 531
(Chan et al., 2010), Jatropha curcas (Sato et al., 2011), Manihot esculenta (Prochnik et al., 532
2012), Cucumis sativus (Huang et al., 2009), Cucumis melo (Garcia-Mas et al., 2012), Carica 533
papaya (Ming et al., 2008), Gossypium raimondii (Wang et al., 2012), and Theobroma cacao 534
(Argout et al., 2011). We also extracted LRR-RLKs from the moss Physcomitrella patens 535
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

26
(Rensing et al., 2008) and the spikemoss Selaginella moellendorffii (Banks et al., 2011). 536
Throughout the article we refer to the species using five-digit identifiers which can be found 537
in Table II. Altogether, we analyzed 33 genomes from 39 proteomes (we used several 538
annotation versions of the A. thaliana and O. sativa genomes). A table containing the details 539
on which genome versions we used can be found in Supplemental Table SV. The phylogeny 540
of those species is provided in Fig. 4. 541
542
LRR-RLK extraction, clustering, phylogeny, and identification of gain/loss events 543
We used the hmmsearch program (Eddy, 2009) to extract peptide sequences containing both, 544
intact (i.e. non-degenerated) LRR(s) and a KD from the proteomes as previously described by 545
(Dievart et al., 2011). We classified SGs using the KD by a global phylogenetic analysis (the 546
tree can be found at http://phylogeny.southgreen.fr/kinase/index.php - Global Analysis). First, 547
sequences were aligned using MAFFT (Katoh et al., 2005) with a progressive strategy. 548
Second, the alignments were cleaned using TrimAl (Capella-Gutiérrez et al., 2009) with 549
settings to remove every site with more than 20% of gaps or with a similarity score lower than 550
0.001. Third, a similarity matrix was computed by ProtDist (Felsenstein, 1993) using a JTT 551
model. Fourth, a global distance phylogeny was inferred using FastME (Desper and Gascuel, 552
2006) with default settings and SPR movements to optimize the tree topology. Fifth, SGs 553
were defined manually in the global phylogeny using the Arabidopsis genes as reference 554
which lead us to 20 SGs in contrast to the 15 previously described (Shiu et al., 2004; Lehti-555
Shiu et al., 2009). 556
More accurate phylogenies were then inferred for each of the 20 SGs. The KD of the 557
sequences attributed to each SG were re-aligned using MAFFT with an iterative strategy 558
(maximum of 100 iterations). Alignments were cleaned using TrimAl with settings to only 559
remove sites with more than 80% of gaps. Then, maximum likelihood phylogenies were 560
inferred by PhyML 3.0 (Guindon et al., 2010) using a LG+gamma model and the best of NNI 561
and SPR topology optimization. Statistical branch support was computed using the aLRT/SH-562
like strategy (Guindon et al. 2010). This left us with 20 phylogenies, one for each SG (all 563
phylogenies are available at http://phylogeny.southgreen.fr/kinase/index.php - SG_I - 564
SG_XV). 565
Each of the 20 phylogenetic trees has been reconciled with the species tree using RAP-566
Green (Dufayard et al., 2005; https://github.com/SouthGreenPlatform/rap-green). By 567
comparing the gene tree to the species tree, this analysis allows to root phylogenetic trees and 568
to infer duplication and loss events (Dufayard et al., 2005). We tested this approach of rooting 569
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

27
(by minimizing the number of inferred duplications and losses) and compared it to rooting 570
with outgroups (data not shown). The two methods provided very close root locations that did 571
not change the overall conclusions. Using this RAP-Green tree reconciliation approach 572
(parameters: Maximum support for reduction 0.95), we inferred the number of duplications 573
and losses at each node of the species tree. Briefly, each duplication and loss respectively 574
increases and decreases by one the number of copies in the common ancestor of the 575
taxonomic group analyzed. 576
We determined the global SG- and species-specific expansion rate by computing the 577
number of LRR-RLK genes in one SG divided by the ancestral number and number of LRR-578
RLK genes in one species divided by the ancestral number, respectively. An ANOVA 579
analysis showed that the expansion rate differed significantly between the SGs/species (p<2e-580
16 in both cases). We used the TukeyHSD test of the agricolae package (http://cran.r-581
project.org/web/packages/agricolae/index.html) in R (R Development Core Team, 2012) to 582
further explore which groups of SGs/species differ from each other. This test compares the 583
range of sample means and defines an Honest Significance Difference (HSD) value which is 584
the minimum distance between groups to be considered statistically significant. In short, 585
TukeyHSD is a post-hoc test which groups subsets by significance levels after ANOVA 586
showed significant differences between subsets. 587
588
LSE dataset and testing for positive selection 589
Testing for adaptation can be done by comparing positive (Darwinian) selection footprints in 590
lineages with recently and specifically duplicated genes to reference lineages containing only 591
single-copy genes. One way to infer positive selection is by analyzing nucleotide substitution 592
data at the codon level in a phylogenetic framework. As nucleotide substitutions can either be 593
nonsynonymous (i.e. protein changing, thereby potentially impacting the fitness) or 594
synonymous (i.e. not protein changing, thereby theoretically without consequences for the 595
fitness, but see (Lawrie et al., 2013)), the nonsynonymous/synonymous substitution rate ratio, 596
denoted as dN/dS or ω, can be used to infer the direction and strength of natural selection. An 597
ω<1 indicates purifying selection and the closer ω is to 0, the stronger purifying selection is 598
acting. Under neutral evolution, ω=1. An ω>1 indicates that positive selection is acting. 599
We identified ultraparalog clusters (UP – only related by duplication) using a tree 600
reconciliation approach (Dufayard et al., 2005). Those represent our LSE gene set. As a 601
single-copy gene reference, we chose a superortholog gene set (SO – only related by 602
speciation). We chose clusters with a minimum of five sequences. To address the question of 603
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

28
whether or not positive selection is more frequent after LSE events, we compared the results 604
obtained on UPs with those obtained on SO gene sets. Species which diverged <15 Mya were 605
merged for the LSE detection (see Fig. 4) in order not to overly reduce the ultraparalog (UP) 606
dataset and to not induce bias due to very recent speciation events: ANDRO (= ZEAMA and 607
SORBI), ORYZA (= ORYSJ and ORYSI), SOLAN (= SOLLC and SOLTU), CUCUM (= 608
CUCSA and CUCME), BRASS (= ARATH, ARALY, BRARA, SCHPA and EUTSA). We 609
then applied the pipeline developed by Fischer et al. (2014) to the extracted UP and SO 610
clusters. In short, the pipeline consists of following steps: (i) The clusters were aligned using 611
PRANK+F with codon option (Löytynoja and Goldman, 2005). The alignments were cleaned 612
by GUIDANCE (Penn et al., 2010) with the default sequence quality cut-off and a column 613
cut-off of 0.97 to remove problematic sequences and unreliable sites from the alignments. We 614
used PRANK and GUIDANCE here as previous benchmarks (Fletcher and Yang, 2010; 615
Jordan and Goldman, 2012) showed that these programs lead to a minimum of false positives 616
when inferring positive selection using codeml. The cleaned alignments can be retrieved here: 617
http://phylogeny.southgreen.fr/kinase/alignments.php – Alignments: Manually curated 618
alignments for positive selection analysis. (ii) We relied on the egglib package (De Mita and 619
Siol, 2012) to infer the maximum likelihood phylogeny at the nucleotide level for every 620
alignment using PhyML 3.0 (Guindon et al., 2010) under the GTR substitution model. (iii) 621
We ran the codeml site model implemented in the PAML4 software (Yang, 2007) to infer 622
positive selection on codons under several substitution models. In clusters identified to have 623
evolved under positive selection, Bayes empirical Bayes was used to calculate the posterior 624
probabilities at each codon and detect those under positive selection (i.e. those with a 625
posterior probability of ω>1 strictly above 95%). All alignments detected to be under positive 626
selection at the codon level were curated manually for potential alignment errors. A table 627
containing the details on all codons showing a signal of positive selection using codeml can 628
be found in Supplemental Table SIII. (iv) We used mapNH (Dutheil et al., 2012; Romiguier et 629
al., 2012) to infer ω at the branch level. 630
In order to analyze the distribution of positively selected sites among domains, we 631
calculated a hit frequency that computes the number of sites under positive selection found in 632
each domain relative to all sites possible. All possible sites for each domain were calculated 633
as follows: First, we extracted the size of each domain of every SG. If SGs were subdivided 634
further we took the average size of each domain. Second, we multiplied the size of each 635
domain by the number of UP clusters we found for each SG. For example: the LRR of SG_I* 636
contains an average of 77 sites. We found 8 UP clusters for SG_I*. Therefore, the total 637
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

29
number of possible LRR sites for SG_I* is 77*8=616 sites. Third, we added the sites for each 638
domain up for all SGs. 639
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

30
SUPPLEMENTAL DATA 640
Supplemental Tables SI – SV can be obtained from: 641
http://phylogeny.southgreen.fr/kinase/index.php - Tables 642
Supplemental Table SI: Estimated divergence times and corresponding references for Fig. 4. 643
Supplemental Table SII: Results of the TukeyHSD test. 644
Supplemental Table SIII: Details on all codons showing a signal of positive selection using 645
codeml. 646
Supplemental Table SIV: Arabidopsis LRR-RLK gene classification according to TAIR 647
(https://www.arabidopsis.org/) 648
Supplemental Table SV: List of genomes used here, with name, link, and version of the 649
genome fasta file. 650
Supplemental Figure S1: Summary of ultraparalog and superortholog cluster size and 651
length. 652
Each dot represents (A) an ultraparalog or (B) a superortholog alignment. The histogram 653
above the scatter plot represents the count of alignments for each cluster size (= number of 654
sequences in alignment); the histogram right to the scatter plot represents the frequency of 655
alignments for each alignment length. 656
Supplemental Figure S2: ω distribution of branches of UP and SO clusters. 657
The distribution of ω of branches in UP (purple) and SO clusters (yellow). 658
659
660
ACKNOWLEGEMENTS 661
The authors thank the reviewers for their helpful comments. 662
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

31
Table I: Total number of LRR-RLK in our angiosperm dataset, number of ancestral genes in 663
LCAA, and median global expansion rate for each SG among the 31 species. 664
SG Total number
of genes Number of
ancestral genes Median global expansion rate
I* 482 7 2.00 II 349 9 1.22
III* 1,400 32 1.22 IV 131 3 1.33 V 263 5 1.80
VI* 324 10 1.00 VIIa 157 3 1.67 VIIb 84 2 1.50
VIII-1 216 5 1.40 VIII-2 355 8 1.25
IX 193 3 1.67 Xa 143 2 2.00
Xb* 367 9 1.11 XI* 1,177 29 1.28 XIIa 1,126 9 3.00 XIIb 423 4 2.00 XIIIa 84 2 1.50 XIIIb 77 2 1.00 XIV 84 1 3.00 XV* 119 5 0.80 Total 7,554 150
665
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

32
Table II: Five-digit code for each species. 666
Species name Common name Five-digit code Phoenix dactylifera Date palm PHODC
Musa acuminata Banana MUSAC
Brachypodium distachyon Purple false brome BRADI
Oryza sativa ssp. japonica Asian rice ORYSJ
Oryza sativa ssp. indica Indian rice ORYSI
Setaria italica Foxtail millet SETIT
Zea mays Maize MAIZE
Sorghum bicolor Milo SORBI
Solanum tuberosum Potato SOLTU
Solanum lycopersicum Tomato SOLLC
Vitis vinifera Common grape vine VITVI
Theobroma cacao Cacao tree THECC
Gossypium raimondii Cotton progenitor GOSRA
Carica papaya Papaya CARPA
Arabidopsis thaliana Thale cress ARATH
Arabidopsis lyrata Out-crossing ARATH relative ARALY
Brassica rapa Turnip BRARA
Schrenkiella parvula A saltwater cress SCHPA
Eutrema salsugineum A saltwater cress EUTSA
Cucumis sativus Cucumber CUCSA
Cucumis melo Melon CUCME
Prunus persica Peach PRUPE
Malus x domestica Apple MALDO
Lotus japonicus LOTJA
Medicago truncatula Barrel medic MEDTR
Glycine max Soybean GLYMA
Cajanus cajan Pigeon pea CAJCA
Populus trichocarpa Black cottonwood POPTR
Ricinus communis Castor oil plant RICCO
Jatropha curcas Barbados nut JATCU
Maniohot esculenta Cassava MANES
Selaginella moellendorffii A spikemoss SELML
Physcomitrella patens A moss PHYPA
667
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

33
Table III: Polyploidy events Estimated times of polyploidy event and corresponding 668
references for Fig. 4. 669
Event Name Reference Age [million years]
1 Seed plant tetraploidy (Jiao et al., 2011) 350-330
2 Angiosperm tetraploidy (Jiao et al., 2011) 230-190
3 Monocot tetraploidy (Tang et al., 2010a) 130
4 Date palm WGD (D'Hont et al., 2012) 75-65 (?)
5 Banana gamma (D'Hont et al., 2012) 100
6 Banana beta (D'Hont et al., 2012) 65
7 Banana alpha (D'Hont et al., 2012) 65
8 Grass tetraploidy B (sigma) (D'Hont et al., 2012) 123-109
9 Grass tetraploidy (rho) (Paterson et al., 2004) 70
10 Maize tetraploidy (Schnable et al., 2011) 12-5
11 Eudicot hexaploidy
(Arabidopsis gamma)
(Jaillon et al., 2007; Cenci et
al., 2010; Wang et al., 2012) 150-120
12 Solanum hexaploidy (The Tomato Genome
Consortium, 2012) 91-52
13 Papiloniod tetraploidy (Pfeil et al., 2005) 55-54
14 Soybean tetraploidy (Pfeil et al., 2005) 15-13
15 Apple tetraploidy (Velasco et al., 2010; Verde
et al., 2013) 45-30
16 Poplar tetraploidy (Tuskan et al., 2006) 65-60
17 Arabidopsis beta (Fawcett et al., 2009) 70-40
18 Arabidopsis alpha (Barker et al., 2009) 23
19 Brassica hexaploidy (Wang et al., 2011) 9-5
20 Cotton WGD (Wang et al., 2012) 20-13
21 Cassava WGD (Mühlhausen and Kollmar,
2013)
? (after
Crotonoideae split)
670
671
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

34
Table IV: Details of the LSE and mapNH analysis for UP and SO clusters. 672
UP SO
total number of clusters 75 189
clusters for final mapNH analysis 71 176
median cluster size (1st; 3rd Qu) 8 (6; 12) 8 (6; 14)
min; max cluster size 5; 38 5; 25
median alignment length (1st; 3rd Qu) 3,237
(2,952; 3,574)
2,841
(2,034; 3,192)
min; max alignment length 1,749; 8,691 861; 6,216
branches analyzed/total number of
branches
1,193/1,246 2,860/2,960
clusters with branches omega > 1.0 (%) 25 (35.21) 10 (5.68)
branches with omega < 1.0 (%) 1,121 (93.96) 2,846 (99.51)
mean omega for < 1.0 branches +/- sd 0.48 +/- 0.17 0.24 +/- 0.12
branches with omega > 1.0 (%) 72 (6.04) 14 (0.49)
mean omega for > 1.0 branches +/- sd 1.45 +/- 0.51 1.13 +/- 0.14
mean omega +/- sd 0.54 +/- 0.31 0.24 +/- 0.13
673
674
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

35
FIGURE LEGENDS 675
Figure 1: Global expansion rate in each subgroup: Total number of genes in each species 676
divided by the ancestral number (see Table I). An ANOVA test showed that the expansion 677
rate differs significantly between SGs (p<2e-16). Therefore, we performed a TukeyHSD test 678
to determine which SGs exactly show a significant difference between each other and group 679
those SGs by significance level (a-e). Letters above the boxplot indicate TukeyHSD 680
significance group (see Supplemental Table SII). The significance groups are color coded 681
according to the mean expansion rate: orange: >2.25 fold expansion; red: 1.75-2.25 fold 682
expansion; purple: 1.30-1.75 fold expansion; blue: 0.75-1.30 fold expansion (i.e. no 683
expansion). The outlier species are labeled for each SG. See Table II for species IDs. 684
685
Figure 2: Branch-specific expansion/diminution of LRR-RLK genes for every SG on every 686
branch in the phylogenetic tree. The tree on the left displays all the nodes and branches, 687
polyploidy events are marked with dots. Every line gives the expansion rate where the current 688
(descendant) node is compared to the previous (ascendant) node. Red boxes indicate 689
expansion, blue boxes indicate diminution, and blank boxes indicate stagnation. For example: 690
SG_I* has the same number of copies in monocots compared to the ascendant node (= 691
angiosperms) indicated by a blank box. In PHODC, a diminution occurred compared to the 692
ascendant node (= monocots) indicated by a blue box. In MUSAC, an expansion occurred 693
compared to the ascendant node (= monocots) indicated by a red box and so on. 694
695
Figure 3: Global expansion rate in each species: Total number of genes in each species 696
divided by the ancestral number (see Table I). An ANOVA test showed that the expansion 697
rate differs significantly between species (p<2e-16). Therefore, we performed a TukeyHSD 698
test to determine which species exactly show a significant difference between each other and 699
group those species by significance level (a-e). Letters above the boxplot indicate TukeyHSD 700
significance group (see Supplemental Table SII). The significance groups are color coded 701
according to the mean expansion rate: orange: >2.25 fold expansion; red: 1.75-2.25 fold 702
expansion; purple: 1.40-1.75 fold expansion; blue: 0.80-1.40 fold expansion (i.e. no 703
expansion). The outlier SGs are labeled for each species. See Table II for species IDs. 704
705
Figure 4: Phylogenetic tree of the 33 species studied here. Five-digit species identifiers are 706
given in parenthesis next to the species name. Species which diverged <15 mya were merged 707
for the LSE analysis (see Materials and Methods): ANDRO, ORYZA, SOLAN, CUCUM, 708
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

36
BRASS. Polyploidy events and their estimated ages are indicated on the tree: circles on the 709
branches represent WGD, dark circles represent WGT. The numbers in the circles refer to 710
details on the polyploidization events given in Table I. Species divergence and their estimated 711
age are indicated by grey squares on the nodes. The numbers in the squares refer to details on 712
the divergence times given in Supplemental Table SI. Dots and asterisks on the branches 713
indicate SG expansions. Dots, 2-fold; small asterisks, between 2 and 4-fold; large asterisks, 714
equal or more than 4-fold. SG I* (brown), SG_IV (dark green), SG_V (grey), SG_VIIa 715
(orange), SG_VIIb (yellow), SG_VIII-1 (dark brown), SG_VIII-2 (green), SG_IX (light blue), 716
SG_Xa (dark blue), SG_XIIa (pink), SG_XIIb (purple), SG_XIIIa (red), SG_XIV (black), 717
SG_XV* (white). The asterisks and dots do not indicate the exact age. 718
719
Figure 5: Distribution of UP and SO clusters and sequences over all SGs. Frequency of all 720
extracted UP (dark blue) and SO clusters (dark orange) for each SG; Frequency of all 721
extracted UP (light blue) and SO sequences (light orange) for each SG. 722
723
Figure 6: (A) Hit frequency (i.e. frequency of codons under selection vs. total number of 724
sites) for each domain of the LRR-RLK genes. (B) Schematic structure of the LRR-RLK 725
genes, here with SG_I* gene structure as an example. The absence/presence and size of the 726
domains varies between SGs, please see text for details. N-term, N-terminal end; SP (dark 727
grey), signal peptide; Cys-pair 1 (blue), first cysteine pair; NC1, N-terminal of Cys-pair 1; 728
CC1, C-terminal of Cys-pair 1; other (green), other domains; LRR (red), leucine-rich repeat; 729
Cys-pair 2 (blue), first cysteine pair; NC2, N-terminal of Cys-pair 2; CC2, C-terminal of Cys-730
pair 2; TM (black), trans-membrane domain; JM, juxta-membrane domain; KD (yellow), 731
kinase domain; C-term, C-terminal end; inter, other inter-domain regions. (C) Frequency of 732
amino acids in the LRR domain under positive selection. L, leucine; N, asparagine; G, 733
glycine; I, isoleucine; P, proline; x, variable; is, island between LRRs. 734
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

37
735
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

Parsed CitationsAhmad R, Parfitt D, Fass J, Ogundiwin E, Dhingra A, Gradziel T, Lin D, Joshi N, Martinez-Garcia P, Crisosto C (2011) Whole genomesequencing of peach (Prunus persica L.) for SNP identification and selection. BMC Genomics 12: 569
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Al-Dous EK, George B, Al-Mahmoud ME, Al-Jaber MY, Wang H, Salameh YM, Al-Azwani EK, Chaluvadi S, Pontaroli AC, DeBarry J,Arondel V, Ohlrogge J, Saie IJ, Suliman-Elmeer KM, Bennetzen JL, Kruegger RR, Malek JA (2011) De novo genome sequencingand comparative genomics of date palm (Phoenix dactylifera). Nat Biotechnol 29: 521-527
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Albert M, Jehle AK, Mueller K, Eisele C, Lipschis M, Felix G (2010) Arabidopsis thaliana pattern recognition receptors for BacterialElongation Factor Tu and Flagellin can be combined to form functional chimeric receptors. J Biol Chem 285: 19035-19042
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Argout X, Salse J, Aury JM, Guiltinan MJ, Droc G, Gouzy J, Allegre M, Chaparro C, Legavre T, Maximova SN, Abrouk M, Murat F,Fouet O, Poulain J, Ruiz M, Roguet Y, Rodier-Goud M, Barbosa-Neto JF, Sabot F, Kudrna D, Ammiraju JS, Schuster SC, CarlsonJE, Sallet E, Schiex T, Dievart A, Kramer M, Gelley L, Shi Z, Berard A, Viot C, Boccara M, Risterucci AM, Guignon V, Sabau X, AxtellMJ, Ma Z, Zhang Y, Brown S, Bourge M, Golser W, Song X, Clement D, Rivallan R, Tahi M, Akaza JM, Pitollat B, Gramacho K,D'Hont A, Brunel D, Infante D, Kebe I, Costet P, Wing R, McCombie WR, Guiderdoni E, Quetier F, Panaud O, Wincker P, Bocs S,Lanaud C (2011) The genome of Theobroma cacao. Nat Genet 43: 101-108
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Arya P, Kumar G, Acharya V, Singh AK (2014) Genome-wide identification and expression analysis of NBS-encoding genes in Malusx domestica and expansion of NBS genes family in Rosaceae. PLOS ONE 9: e107987
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Banks JA, Nishiyama T, Hasebe M, Bowman JL, Gribskov M, dePamphilis C, Albert VA, Aono N, Aoyama T, Ambrose BA, Ashton NW,Axtell MJ, Barker E, Barker MS, Bennetzen JL, Bonawitz ND, Chapple C, Cheng C, Correa LGG, Dacre M, DeBarry J, Dreyer I,Elias M, Engstrom EM, Estelle M, Feng L, Finet C, Floyd SK, Frommer WB, Fujita T, Gramzow L, Gutensohn M, Harholt J, Hattori M,Heyl A, Hirai T, Hiwatashi Y, Ishikawa M, Iwata M, Karol KG, Koehler B, Kolukisaoglu U, Kubo M, Kurata T, Lalonde S, Li K, Li Y, LittA, Lyons E, Manning G, Maruyama T, Michael TP, Mikami K, Miyazaki S, Morinaga Si, Murata T, Mueller-Roeber B, Nelson DR,Obara M, Oguri Y, Olmstead RG, Onodera N, Petersen BL, Pils B, Prigge M, Rensing SA, Riaño-Pachón DM, Roberts AW, Sato Y,Scheller HV, Schulz B, Schulz C, Shakirov EV, Shibagaki N, Shinohara N, Shippen DE, Sørensen I, Sotooka R, Sugimoto N, SugitaM, Sumikawa N, Tanurdzic M, Theißen G, Ulvskov P, Wakazuki S, Weng JK, Willats WWGT, Wipf D, Wolf PG, Yang L, Zimmer AD,Zhu Q, Mitros T, Hellsten U, Loqué D, Otillar R, Salamov A, Schmutz J, Shapiro H, Lindquist E, Lucas S, Rokhsar D, Grigoriev IV(2011) The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 332: 960-963
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Barker MS, Vogel H, Schranz ME (2009) Paleopolyploidy in the Brassicales: Analyses of the Cleome transcriptome elucidate thehistory of genome duplications in Arabidopsis and other Brassicales. Genome Biol Evol 1: 391-399
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Bowers JE, Chapman BA, Rong J, Paterson AH (2003) Unravelling angiosperm genome evolution by phylogenetic analysis ofchromosomal duplication events. Nature 422: 433-438
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Cannon SB, McKain MR, Harkess A, Nelson MN, Dash S, Deyholos MK, Peng Y, Joyce B, Stewart CN, Rolf M, Kutchan T, Tan X,Chen C, Zhang Y, Carpenter E, Wong GK-S, Doyle JJ, Leebens-Mack J (2014) Multiple polyploidy events in the early radiation ofnodulating and non-nodulating legumes. Mol Biol Evol: doi:10.1093/molbev/msu1296
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scalephylogenetic analyses. Bioinformatics 25: 1972-1973
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Castells E, Casacuberta JM (2007) Signalling through kinase-defective domains: the prevalence of atypical receptor-like kinases inplants. J Exp Bot 58: 3503-3511
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Cenci A, Combes M-C, Lashermes P (2010) Comparative sequence analyses indicate that Coffea (Asterids) and Vitis (Rosids)derive from the same paleo-hexaploid ancestral genome. Mol Genet Genomics 283: 493-501
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J, Puiu D, Melake-Berhan A, Jones KM, Redman J, Chen G, Cahoon EB, Gedil M,Stanke M, Haas BJ, Wortman JR, Fraser-Liggett CM, Ravel J, Rabinowicz PD (2010) Draft genome sequence of the oilseed speciesRicinus communis. Nat Biotechnol 28: 951-956
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Chevalier D, Batoux M, Fulton L, Pfister K, Yadav RK, Schellenberg M, Schneitz K (2005) STRUBBELIG defines a receptor kinase-mediated signaling pathway regulating organ development in Arabidopsis. Proc Natl Acad Sci USA 102: 9074-9079
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
D'Hont A, Denoeud F, Aury JM, Baurens FC, Carreel F, Garsmeur O, Noel B, Bocs S, Droc G, Rouard M, Da Silva C, Jabbari K,Cardi C, Poulain J, Souquet M, Labadie K, Jourda C, Lengelle J, Rodier-Goud M, Alberti A, Bernard M, Correa M, Ayyampalayam S,McKain MR, Leebens-Mack J, Burgess D, Freeling M, Mbeguie AMD, Chabannes M, Wicker T, Panaud O, Barbosa J, Hribova E,Heslop-Harrison P, Habas R, Rivallan R, Francois P, Poiron C, Kilian A, Burthia D, Jenny C, Bakry F, Brown S, Guignon V, Kema G,Dita M, Waalwijk C, Joseph S, Dievart A, Jaillon O, Leclercq J, Argout X, Lyons E, Almeida A, Jeridi M, Dolezel J, Roux N, RisterucciAM, Weissenbach J, Ruiz M, Glaszmann JC, Quetier F, Yahiaoui N, Wincker P (2012) The banana (Musa acuminata) genome and theevolution of monocotyledonous plants. Nature 488: 213-217
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Dassanayake M, Oh DH, Haas JS, Hernandez A, Hong H, Ali S, Yun DJ, Bressan RA, Zhu JK, Bohnert HJ, Cheeseman JM (2011)The genome of the extremophile crucifer Thellungiella parvula. Nat Genet 43: 913-918
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
De Mita S, Siol M (2012) EggLib: processing, analysis and simulation tools for population genetics and genomics. BMC Genet 13:27
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
De Smet I, Voss U, Jurgens G, Beeckman T (2009) Receptor-like kinases shape the plant. Nat Cell Biol 11: 1166-1173Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
De Smet R, Adams KL, Vandepoele K, Van Montagu MCE, Maere S, Van de Peer Y (2013) Convergent gene loss following geneand genome duplications creates single-copy families in flowering plants. Proc Natl Acad Sci USA 110: 2898-2903
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Desper R, Gascuel O (2006) Getting a Tree Fast: Neighbor Joining, FastME, and Distance-Based Methods. In I John Wiley & Sons,ed, Curr Protoc Bioinformatics, pp 6.3.1-6.3.28
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Dievart A, Gilbert N, Droc G, Attard A, Gourgues M, Guiderdoni E, Perin C (2011) Leucine-rich repeat receptor kinases aresporadically distributed in eukaryotic genomes. BMC Evol Biol 11: 367
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Dodds PN, Rathjen JP (2010) Plant immunity: towards an integrated view of plant-pathogen interactions. Nat Rev Genet 11: 539-548Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Dou L, Zhang X, Pang C, Song M, Wei H, Fan S, Yu S (2014) Genome-wide analysis of the WRKY gene family in cotton. Mol GenetGenomics 289: 1103-1121
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

Dufayard JF, Duret L, Penel S, Gouy M, Rechenmann F, Perrière G (2005) Tree pattern matching in phylogenetic trees: automaticsearch for orthologs or paralogs in homologous gene sequence databases. Bioinformatics 21: 2596-2603
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Dutheil JY, Galtier N, Romiguier J, Douzery EJ, Ranwez V, Boussau B (2012) Efficient selection of branch-specific models ofsequence evolution. Mol Biol Evol 29: 1861-1874
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Eddy SR (2009) A new generation of homology search tools based on probabilistic inference. Genome Inform October 2009: 205-211
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Enkhbayar P, Kamiya M, Osaki M, Matsumoto T, Matsushima N (2004) Structural principles of leucine-rich repeat (LRR) proteins.Proteins: Struct, Funct, Bioinf 54: 394-403
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Fawcett JA, Maere S, Van de Peer Y (2009) Plants with double genomes might have had a better chance to survive theCretaceous-Tertiary extinction event. Proc Natl Acad Sci USA 106: 5737-5742
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Felsenstein J (1993) PHYLIP (Phylogeny Inference Package) version 3.5c. Distributed by the author, Department of Genetics,University of Washington, Seattle
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Fischer I, Dainat J, Ranwez V, Glémin S, Dufayard J-F, Chantret N (2014) Impact of recurrent gene duplication on adaptation ofplant genomes. BMC Plant Biol 14: 151
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Fletcher W, Yang Z (2010) The effect of insertions, deletions, and alignment errors on the branch-site test of positive selection.Mol Biol Evol 27: 2257-2267
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Garcia-Mas J, Benjak A, Sanseverino W, Bourgeois M, Mir G, González VM, Hénaff E, Câmara F, Cozzuto L, Lowy E, Alioto T,Capella-Gutiérrez S, Blanca J, Cañizares J, Ziarsolo P, Gonzalez-Ibeas D, Rodríguez-Moreno L, Droege M, Du L, Alvarez-TejadoM, Lorente-Galdos B, Melé M, Yang L, Weng Y, Navarro A, Marques-Bonet T, Aranda MA, Nuez F, Picó B, Gabaldón T, Roma G,Guigó R, Casacuberta JM, Arús P, Puigdomènech P (2012) The genome of melon (Cucumis melo L.). Proc Natl Acad Sci USA 109:11872-11877
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Gish LA, Clark SE (2011), The RLK/Pelle family of kinases. Plant J 66: 117-127Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59: 307-321
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Hahn MW (2009) Distinguishing among evolutionary models for the maintenance of gene duplicates. J Hered 100: 605-617Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Han MV, Thomas GWC, Lugo-Martinez J, Hahn MW (2013) Estimating gene gain and loss rates in the presence of error in genomeassembly and annotation using CAFE 3. Mol Biol Evol 30: 1987-1997
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Hanada K, Zou C, Lehti-Shiu MD, Shinozaki K, Shiu SH (2008) Importance of lineage-specific expansion of plant tandem duplicates www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from
Copyright © 2016 American Society of Plant Biologists. All rights reserved.

in the adaptive response to environmental stimuli. Plant Physiol 148: 993-1003Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Hok S, Danchin EGJ, Allasia V, Panabières F, Attard A, Keller H (2011) An Arabidopsis (malectin-like) leucine-rich repeat receptor-like kinase contributes to downy mildew disease. Plant, Cell Environ 34: 1944-1957
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Hu TT, Pattyn P, Bakker EG, Cao J, Cheng JF, Clark RM, Fahlgren N, Fawcett JA, Grimwood J, Gundlach H, Haberer G, HollisterJD, Ossowski S, Ottilar RP, Salamov AA, Schneeberger K, Spannagl M, Wang X, Yang L, Nasrallah ME, Bergelson J, Carrington JC,Gaut BS, Schmutz J, Mayer KF, Van de Peer Y, Grigoriev IV, Nordborg M, Weigel D, Guo YL (2011) The Arabidopsis lyrata genomesequence and the basis of rapid genome size change. Nat Genet 43: 476-481
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, Lucas WJ, Wang X, Xie B, Ni P, Ren Y, Zhu H, Li J, Lin K, Jin W, Fei Z, Li G, Staub J, KilianA, van der Vossen EA, Wu Y, Guo J, He J, Jia Z, Ren Y, Tian G, Lu Y, Ruan J, Qian W, Wang M, Huang Q, Li B, Xuan Z, Cao J, Asan,Wu Z, Zhang J, Cai Q, Bai Y, Zhao B, Han Y, Li Y, Li X, Wang S, Shi Q, Liu S, Cho WK, Kim JY, Xu Y, Heller-Uszynska K, Miao H,Cheng Z, Zhang S, Wu J, Yang Y, Kang H, Li M, Liang H, Ren X, Shi Z, Wen M, Jian M, Yang H, Zhang G, Yang Z, Chen R, Liu S, Li J,Ma L, Liu H, Zhou Y, Zhao J, Fang X, Li G, Fang L, Li Y, Liu D, Zheng H, Zhang Y, Qin N, Li Z, Yang G, Yang S, Bolund L, KristiansenK, Zheng H, Li S, Zhang X, Yang H, Wang J, Sun R, Zhang B, Jiang S, Wang J, Du Y, Li S (2009) The genome of the cucumber,Cucumis sativus L. Nat Genet 41: 1275-1281
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Innan H (2009) Population genetic models of duplicated genes. Genetica 137: 19-37Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Innan H, Kondrashov F (2010) The evolution of gene duplications: classifying and distinguishing between models. Nat Rev Genet11: 97-108
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436: 793-800Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F,Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyere C, Billault A, Segurens B, Gouyvenoux M, Ugarte E,Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M,Scalabrin S, Canaguier A, Le Clainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M,Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Pe ME, Valle G, Morgante M, Caboche M, Adam-Blondon AF, Weissenbach J,Quetier F, Wincker P, French-Italian Public Consortium for Grapevine Genome C (2007) The grapevine genome sequencesuggests ancestral hexaploidization in major angiosperm phyla. Nature 449: 463-467
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, Soltis DE, CliftonSW, Schlarbaum SE, Schuster SC, Ma H, Leebens-Mack J, dePamphilis CW (2011) Ancestral polyploidy in seed plants andangiosperms. Nature 473: 97-100
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Johnson DA, Thomas MA (2007) The monosaccharide transporter gene family in Arabidopsis and rice: a history of duplications,adaptive evolution, and functional divergence. Mol Biol Evol 24: 2412-2423
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Jones DA, Jones JDG (1997) The role of leucine-rich repeat proteins in plant defences. Adv Bot Res 24: 90-167Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Jordan G, Goldman N (2012) The effects of alignment error and alignment filtering on the sitewise detection of positive selection.Mol Biol Evol 29: 1125-1139
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from
Copyright © 2016 American Society of Plant Biologists. All rights reserved.

Katoh K, Kuma K-I, Toh H, Miyata T (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. NucleicAcids Res 33: 511-518
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Langham RJ, Walsh J, Dunn M, Ko C, Goff SA, Freeling M (2004) Genomic duplication, fractionation and the origin of regulatorynovelty. Genetics 166: 935-945
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Lawrie DS, Messer PW, Hershberg R, Petrov DA (2013) Strong purifying selection at synonymous sites in D. melanogaster. PLOSGenet 9: e1003527
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Lehti-Shiu M, Zou C, Shiu S-H (2012) Origin, diversity, expansion history, and functional evolution of the plant Receptor-LikeKinase/Pelle family. In F Tax, B Kemmerling, eds, Receptor-like Kinases in Plants, Vol 13. Springer Berlin Heidelberg, pp 1-22
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Lehti-Shiu MD, Zou C, Hanada K, Shiu S-H (2009) Evolutionary history and stress regulation of plant Receptor-Like Kinase/Pellegenes. Plant Physiol 150: 12-26
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Leitch AR, Leitch IJ (2012) Ecological and genetic factors linked to contrasting genome dynamics in seed plants. New Phytol 194:629-646
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Lindner H, Müller LM, Boisson-Dernier A, Grossniklaus U (2012) CrRLK1L receptor-like kinases: not just another brick in the wall.Curr Opin Plant Biol 15: 659-669
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Löytynoja A, Goldman N (2005) An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad SciUSA 102: 10557-10562
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Markmann K, Giczey G, Parniske M (2008) Functional adaptation of a plant Receptor- Kinase paved the way for the evolution ofintracellular root symbioses with bacteria. PLOS Biol 6: e68
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, Senin P, Wang W, Ly BV, Lewis KL, Salzberg SL, Feng L, Jones MR,Skelton RL, Murray JE, Chen C, Qian W, Shen J, Du P, Eustice M, Tong E, Tang H, Lyons E, Paull RE, Michael TP, Wall K, Rice DW,Albert H, Wang ML, Zhu YJ, Schatz M, Nagarajan N, Acob RA, Guan P, Blas A, Wai CM, Ackerman CM, Ren Y, Liu C, Wang J, Wang J,Na JK, Shakirov EV, Haas B, Thimmapuram J, Nelson D, Wang X, Bowers JE, Gschwend AR, Delcher AL, Singh R, Suzuki JY,Tripathi S, Neupane K, Wei H, Irikura B, Paidi M, Jiang N, Zhang W, Presting G, Windsor A, Navajas-Perez R, Torres MJ, Feltus FA,Porter B, Li Y, Burroughs AM, Luo MC, Liu L, Christopher DA, Mount SM, Moore PH, Sugimura T, Jiang J, Schuler MA, Friedman V,Mitchell-Olds T, Shippen DE, dePamphilis CW, Palmer JD, Freeling M, Paterson AH, Gonsalves D, Wang L, Alam M (2008) The draftgenome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452: 991-996
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Moore RC, Purugganan MD (2005) The evolutionary dynamics of plant duplicate genes. Curr Opin Plant Biol 8: 122-128Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Mühlhausen S, Kollmar M (2013) Whole genome duplication events in plant evolution reconstructed and predicted using myosinmotor proteins. BMC Evol Biol 13: 202
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Nei M, Rooney AP (2005) Concerted and birth-and-death evolution of multigene families. Ann Rev Genet 39: 121-152Pubmed: Author and Title
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

CrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Oh D-H, Hong H, Lee SY, Yun D-J, Bohnert HJ, Dassanayake M (2014) Genome structures and transcriptomes signify nicheadaptation for the multiple-ion-tolerant extremophyte Schrenkiella parvula. Plant Physiol 164: 2123-2138
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Oh M-H, Wang X, Kota U, Goshe MB, Clouse SD, Huber SC (2009) Tyrosine phosphorylation of the BRI1 receptor kinase emergesas a component of brassinosteroid signaling in Arabidopsis. Proc Natl Acad Sci USA 106: 658-663
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Parniske M, Hammond-Kosack KE, Golstein C, Thomas CM, Jones DA, Harrison K, Wulff BBH, Jones JDG (1997) Novel diseaseresistance specificities result from sequence exchange between tandemly repeated genes at the Cf-4/9 locus of tomato. Cell 91:821-832
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A,Schmutz J, Spannagl M, Tang H, Wang X, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA,Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang L, Carpita NC, Freeling M, Gingle AR, Hash CT, KellerB, Klein P, Kresovich S, McCann MC, Ming R, Peterson DG, Mehboob ur R, Ware D, Westhoff P, Mayer KF, Messing J, Rokhsar DS(2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457: 551-556
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Paterson AH, Bowers JE, Chapman BA (2004) Ancient polyploidization predating divergence of the cereals, and its consequencesfor comparative genomics. Proc Natl Acad Sci USA 101: 9903-9908
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Penn O, Privman E, Landan G, Graur D, Pupko T (2010) An alignment confidence score capturing robustness to guide treeuncertainty. Mol Biol Evol 27: 1759-1767
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Pfeil BE, Schlueter JA, Shoemaker RC, Doyle JJ (2005) Placing paleopolyploidy in relation to taxon divergence: A phylogeneticanalysis in legumes using 39 gene families. Syst Biol 54: 441-454
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Potato Genome Sequencing Consortium (2011) Genome sequence and analysis of the tuber crop potato. Nature 475: 189-195Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Prochnik S, Marri P, Desany B, Rabinowicz P, Kodira C, Mohiuddin M, Rodriguez F, Fauquet C, Tohme J, Harkins T, Rokhsar D,Rounsley S (2012) The cassava genome: Current progress, future directions. Tropical Plant Biology 5: 88-94
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
R Development Core Team (2012) R: A language and environment for statistical computing. R Foundation for StatisticalComputing, Vienna, Austria
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Renny-Byfield S, Wendel JF (2014) Doubling down on genomes: Polyploidy and crop plants. Am J Bot 101: 1711-1725Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, Shapiro H, Nishiyama T, Perroud P-F, Lindquist EA, Kamisugi Y, Tanahashi T,Sakakibara K, Fujita T, Oishi K, Shin-I T, Kuroki Y, Toyoda A, Suzuki Y, Hashimoto S-i, Yamaguchi K, Sugano S, Kohara Y, FujiyamaA, Anterola A, Aoki S, Ashton N, Barbazuk WB, Barker E, Bennetzen JL, Blankenship R, Cho SH, Dutcher SK, Estelle M, Fawcett JA,Gundlach H, Hanada K, Heyl A, Hicks KA, Hughes J, Lohr M, Mayer K, Melkozernov A, Murata T, Nelson DR, Pils B, Prigge M,Reiss B, Renner T, Rombauts S, Rushton PJ, Sanderfoot A, Schween G, Shiu S-H, Stueber K, Theodoulou FL, Tu H, Van de PeerY, Verrier PJ, Waters E, Wood A, Yang L, Cove D, Cuming AC, Hasebe M, Lucas S, Mishler BD, Reski R, Grigoriev IV, Quatrano RS,Boore JL (2008) The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 319: 64-69
Pubmed: Author and TitleCrossRef: Author and Title www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from
Copyright © 2016 American Society of Plant Biologists. All rights reserved.

Google Scholar: Author Only Title Only Author and Title
Rizzon C, Ponger L, Gaut BS (2006) Striking similarities in the genomic distribution of tandemly arrayed genes in Arabidopsis andrice. PLOS Comput Biol 2: e115
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Romiguier J, Figuet E, Galtier N, Douzery EJ, Boussau B, Dutheil JY, Ranwez V (2012) Fast and robust characterization of time-heterogeneous sequence evolutionary processes using substitution mapping. PLOS ONE 7: e33852
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Sato S, Hirakawa H, Isobe S, Fukai E, Watanabe A, Kato M, Kawashima K, Minami C, Muraki A, Nakazaki N, Takahashi C, NakayamaS, Kishida Y, Kohara M, Yamada M, Tsuruoka H, Sasamoto S, Tabata S, Aizu T, Toyoda A, Shin-i T, Minakuchi Y, Kohara Y, FujiyamaA, Tsuchimoto S, Kajiyama Si, Makigano E, Ohmido N, Shibagaki N, Cartagena JA, Wada N, Kohinata T, Atefeh A, Yuasa S,Matsunaga S, Fukui K (2011) Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res 18: 65-76
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Sato S, Nakamura Y, Kaneko T, Asamizu E, Kato T, Nakao M, Sasamoto S, Watanabe A, Ono A, Kawashima K, Fujishiro T, Katoh M,Kohara M, Kishida Y, Minami C, Nakayama S, Nakazaki N, Shimizu Y, Shinpo S, Takahashi C, Wada T, Yamada M, Ohmido N,Hayashi M, Fukui K, Baba T, Nakamichi T, Mori H, Tabata S (2008) Genome structure of the legume, Lotus japonicus. DNA Res 15:227-239
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD,Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, BarryK, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang XC, Shinozaki K, Nguyen HT,Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA (2010) Genome sequence of thepalaeopolyploid soybean. Nature 463: 178-183
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Schnable JC, Springer NM, Freeling M (2011) Differentiation of the maize subgenomes by genome dominance and both ancientand ongoing gene loss. Proc Natl Acad Sci USA 108: 4069-4074
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, Minx P, Reily AD, CourtneyL, Kruchowski SS, Tomlinson C, Strong C, Delehaunty K, Fronick C, Courtney B, Rock SM, Belter E, Du F, Kim K, Abbott RM,Cotton M, Levy A, Marchetto P, Ochoa K, Jackson SM, Gillam B, Chen W, Yan L, Higginbotham J, Cardenas M, Waligorski J,Applebaum E, Phelps L, Falcone J, Kanchi K, Thane T, Scimone A, Thane N, Henke J, Wang T, Ruppert J, Shah N, Rotter K,Hodges J, Ingenthron E, Cordes M, Kohlberg S, Sgro J, Delgado B, Mead K, Chinwalla A, Leonard S, Crouse K, Collura K, KudrnaD, Currie J, He R, Angelova A, Rajasekar S, Mueller T, Lomeli R, Scara G, Ko A, Delaney K, Wissotski M, Lopez G, Campos D,Braidotti M, Ashley E, Golser W, Kim H, Lee S, Lin J, Dujmic Z, Kim W, Talag J, Zuccolo A, Fan C, Sebastian A, Kramer M, Spiegel L,Nascimento L, Zutavern T, Miller B, Ambroise C, Muller S, Spooner W, Narechania A, Ren L, Wei S, Kumari S, Faga B, Levy MJ,McMahan L, Van Buren P, Vaughn MW, Ying K, Yeh CT, Emrich SJ, Jia Y, Kalyanaraman A, Hsia AP, Barbazuk WB, Baucom RS,Brutnell TP, Carpita NC, Chaparro C, Chia JM, Deragon JM, Estill JC, Fu Y, Jeddeloh JA, Han Y, Lee H, Li P, Lisch DR, Liu S, Liu Z,Nagel DH, McCann MC, SanMiguel P, Myers AM, Nettleton D, Nguyen J, Penning BW, Ponnala L, Schneider KL, Schwartz DC,Sharma A, Soderlund C, Springer NM, Sun Q, Wang H, Waterman M, Westerman R, Wolfgruber TK, Yang L, Yu Y, Zhang L, Zhou S,Zhu Q, Bennetzen JL, Dawe RK, Jiang J, Jiang N, Presting GG, Wessler SR, Aluru S, Martienssen RA, Clifton SW, McCombie WR,Wing RA, Wilson RK (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326: 1112-1115
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Shang H, Li W, Zou C, Yuan Y (2013) Analyses of the NAC transcription factor gene family in Gossypium raimondii Ulbr.:Chromosomal location, structure, phylogeny, and expression patterns. J Integr Plant Biol 55: 663-676
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Shiu S-H, Bleecker AB (2001) Plant Receptor-Like Kinase gene family: Diversity, function, and signaling. Sci. STKE 2001: re22-Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Shiu S-H, Karlowski WM, Pan R, Tzeng Y-H, Mayer KFX, Li W-H (2004) Comparative analysis of the Receptor-Like Kinase family inArabidopsis and rice. Plant Cell 16: 1220-1234
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

Soltis DE, Albert VA, Leebens-Mack J, Bell CD, Paterson AH, Zheng C, Sankoff D, dePamphilis CW, Wall PK, Soltis PS (2009)Polyploidy and angiosperm diversification. Am J Bot 96: 336-348
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Soltis DE, Burleigh JG (2009) Surviving the K-T mass extinction: New perspectives of polyploidization in angiosperms. Proc NatlAcad Sci USA 106: 5455-5456
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Song X, Guo P, Li C, Liu C-M (2010) The cysteine pairs in CLV2 are not necessary for sensing the CLV3 peptide in shoot and rootmeristems. J Integr Plant Biol 52: 774-781
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Sun W, Cao Y, Jansen Labby K, Bittel P, Boller T, Bent AF (2012) Probing the Arabidopsis Flagellin Receptor: FLS2-FLS2association and the contributions of specific domains to signaling function. Plant Cell 24: 1096-1113
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Tan S, Wang D, Ding J, Tian D, Zhang X, Yang S (2011) Adaptive evolution of Xa21 homologs in Gramineae. Genetica 139: 1465-1475
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Tang H, Bowers JE, Wang X, Paterson AH (2010a) Angiosperm genome comparisons reveal early polyploidy in the monocotlineage. Proc Natl Acad Sci USA 107: 472-477
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Tang P, Zhang Y, Sun X, Tian D, Yang S, Ding J (2010b) Disease resistance signature of the leucine-rich repeat receptor-likekinase genes in four plant species. Plant Sci 179: 399-406
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature408: 796-815
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
The International Brachypodium Initiative (2010) Genome sequencing and analysis of the model grass Brachypodium distachyon.Nature 463: 763-768
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
The Tomato Genome Consortium (2012) The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485:635-641
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J,Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M,Chapman J, Chen GL, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q, Cunningham R, Davis J, Degroeve S, Dejardin A,Depamphilis C, Detter J, Dirks B, Dubchak I, Duplessis S, Ehlting J, Ellis B, Gendler K, Goodstein D, Gribskov M, Grimwood J,Groover A, Gunter L, Hamberger B, Heinze B, Helariutta Y, Henrissat B, Holligan D, Holt R, Huang W, Islam-Faridi N, Jones S,Jones-Rhoades M, Jorgensen R, Joshi C, Kangasjarvi J, Karlsson J, Kelleher C, Kirkpatrick R, Kirst M, Kohler A, Kalluri U, LarimerF, Leebens-Mack J, Leple JC, Locascio P, Lou Y, Lucas S, Martin F, Montanini B, Napoli C, Nelson DR, Nelson C, Nieminen K,Nilsson O, Pereda V, Peter G, Philippe R, Pilate G, Poliakov A, Razumovskaya J, Richardson P, Rinaldi C, Ritland K, Rouze P,Ryaboy D, Schmutz J, Schrader J, Segerman B, Shin H, Siddiqui A, Sterky F, Terry A, Tsai CJ, Uberbacher E, Unneberg P, Vahala J,Wall K, Wessler S, Yang G, Yin T, Douglas C, Marra M, Sandberg G, Van de Peer Y, Rokhsar D (2006) The genome of blackcottonwood, Populus trichocarpa (Torr. & Gray). Science 313: 1596-1604
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Varshney RK, Chen W, Li Y, Bharti AK, Saxena RK, Schlueter JA, Donoghue MT, Azam S, Fan G, Whaley AM, Farmer AD, SheridanJ, Iwata A, Tuteja R, Penmetsa RV, Wu W, Upadhyaya HD, Yang SP, Shah T, Saxena KB, Michael T, McCombie WR, Yang B, ZhangG, Yang H, Wang J, Spillane C, Cook DR, May GD, Xu X, Jackson SA (2012) Draft genome sequence of pigeonpea (Cajanus cajan),an orphan legume crop of resource-poor farmers. Nat Biotechnol 30: 83-89
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.

Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, Fontana P, Bhatnagar SK, Troggio M, Pruss D, Salvi S,Pindo M, Baldi P, Castelletti S, Cavaiuolo M, Coppola G, Costa F, Cova V, Dal Ri A, Goremykin V, Komjanc M, Longhi S, Magnago P,Malacarne G, Malnoy M, Micheletti D, Moretto M, Perazzolli M, Si-Ammour A, Vezzulli S, Zini E, Eldredge G, Fitzgerald LM, Gutin N,Lanchbury J, Macalma T, Mitchell JT, Reid J, Wardell B, Kodira C, Chen Z, Desany B, Niazi F, Palmer M, Koepke T, Jiwan D,Schaeffer S, Krishnan V, Wu C, Chu VT, King ST, Vick J, Tao Q, Mraz A, Stormo A, Stormo K, Bogden R, Ederle D, Stella A,Vecchietti A, Kater MM, Masiero S, Lasserre P, Lespinasse Y, Allan AC, Bus V, Chagne D, Crowhurst RN, Gleave AP, Lavezzo E,Fawcett JA, Proost S, Rouze P, Sterck L, Toppo S, Lazzari B, Hellens RP, Durel CE, Gutin A, Bumgarner RE, Gardiner SE, SkolnickM, Egholm M, Van de Peer Y, Salamini F, Viola R (2010) The genome of the domesticated apple (Malus x domestica Borkh.). NatGenet 42: 833-839
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, Zhebentyayeva T, Dettori MT, Grimwood J, Cattonaro F, Zuccolo A,Rossini L, Jenkins J, Vendramin E, Meisel LA, Decroocq V, Sosinski B, Prochnik S, Mitros T, Policriti A, Cipriani G, Dondini L,Ficklin S, Goodstein DM, Xuan P, Fabbro CD, Aramini V, Copetti D, Gonzalez S, Horner DS, Falchi R, Lucas S, Mica E, Maldonado J,Lazzari B, Bielenberg D, Pirona R, Miculan M, Barakat A, Testolin R, Stella A, Tartarini S, Tonutti P, Arus P, Orellana A, Wells C,Main D, Vizzotto G, Silva H, Salamini F, Schmutz J, Morgante M, Rokhsar DS (2013) The high-quality draft genome of peach(Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet 45: 487-494
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Vetter MM, Kronholm I, He F, Haweker H, Reymond M, Bergelson J, Robatzek S, de Meaux J (2012) Flagellin perception variesquantitatively in Arabidopsis thaliana and its relatives. Mol Biol Evol 29: 1655-1667
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Wang G-L, Ruan D-L, Song W-Y, Sideris S, Chen L, Pi L-Y, Zhang S, Zhang Z, Fauquet C, Gaut BS, Whalen MC, Ronald PC (1998)Xa21D encodes a receptor-like molecule with a Leucine-Rich Repeat domain that determines race-specific recognition and issubject to adaptive evolution. Plant Cell 10: 765-779
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Wang K, Wang Z, Li F, Ye W, Wang J, Song G, Yue Z, Cong L, Shang H, Zhu S, Zou C, Li Q, Yuan Y, Lu C, Wei H, Gou C, Zheng Z, YinY, Zhang X, Liu K, Wang B, Song C, Shi N, Kohel RJ, Percy RG, Yu JZ, Zhu YX, Wang J, Yu S (2012) The draft genome of a diploidcotton Gossypium raimondii. Nat Genet 44: 1098-1103
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, Bai Y, Mun JH, Bancroft I, Cheng F, Huang S, Li X, Hua W, Wang J, Wang X, FreelingM, Pires JC, Paterson AH, Chalhoub B, Wang B, Hayward A, Sharpe AG, Park BS, Weisshaar B, Liu B, Li B, Liu B, Tong C, Song C,Duran C, Peng C, Geng C, Koh C, Lin C, Edwards D, Mu D, Shen D, Soumpourou E, Li F, Fraser F, Conant G, Lassalle G, King GJ,Bonnema G, Tang H, Wang H, Belcram H, Zhou H, Hirakawa H, Abe H, Guo H, Wang H, Jin H, Parkin IA, Batley J, Kim JS, Just J, Li J,Xu J, Deng J, Kim JA, Li J, Yu J, Meng J, Wang J, Min J, Poulain J, Wang J, Hatakeyama K, Wu K, Wang L, Fang L, Trick M, LinksMG, Zhao M, Jin M, Ramchiary N, Drou N, Berkman PJ, Cai Q, Huang Q, Li R, Tabata S, Cheng S, Zhang S, Zhang S, Huang S, SatoS, Sun S, Kwon SJ, Choi SR, Lee TH, Fan W, Zhao X, Tan X, Xu X, Wang Y, Qiu Y, Yin Y, Li Y, Du Y, Liao Y, Lim Y, Narusaka Y, WangY, Wang Z, Li Z, Wang Z, Xiong Z, Zhang Z, Brassica rapa Genome Sequencing Project C (2011) The genome of the mesopolyploidcrop species Brassica rapa. Nat Genet 43: 1035-1039
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Wu H-J, Zhang Z, Wang J-Y, Oh D-H, Dassanayake M, Liu B, Huang Q, Sun H-X, Xia R, Wu Y, Wang Y-N, Yang Z, Liu Y, Zhang W,Zhang H, Chu J, Yan C, Fang S, Zhang J, Wang Y, Zhang F, Wang G, Lee SY, Cheeseman JM, Yang B, Li B, Min J, Yang L, Wang J,Chu C, Chen S-Y, Bohnert HJ, Zhu J-K, Wang X-J, Xie Q (2012) Insights into salt tolerance from the genome of Thellungiellasalsuginea. Proc Natl Acad Sci USA 109: 12219-12224
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Xue Z, Duan L, Liu D, Guo J, Ge S, Dicks J, ÓMáille P, Osbourn A, Qi X (2012) Divergent evolution of oxidosqualene cyclases inplants. New Phytol 193: 1022-1038
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Yang T, Chaudhuri S, Yang L, Du L, Poovaiah BW (2010) A calcium/calmodulin-regulated member of the Receptor-like Kinase familyconfers cold tolerance in plants. J Biol Chem 285: 7119-7126
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from
Copyright © 2016 American Society of Plant Biologists. All rights reserved.

Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24: 1586-1591Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Yang Z, Wang Y, Zhou Y, Gao Q, Zhang E, Zhu L, Hu Y, Xu C (2013a) Evolution of land plant genes encoding L-Ala-D/L-Gluepimerases (AEEs) via horizontal gene transfer and positive selection. BMC Plant Biol 13: 34
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Yang ZL, Liu HJ, Wang XR, Zeng QY (2013b) Molecular evolution and expression divergence of the Populus polygalacturonasesupergene family shed light on the evolution of increasingly complex organs in plants. New Phytol 197: 1353-1365
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Young ND, Debelle F, Oldroyd GE, Geurts R, Cannon SB, Udvardi MK, Benedito VA, Mayer KF, Gouzy J, Schoof H, Van de Peer Y,Proost S, Cook DR, Meyers BC, Spannagl M, Cheung F, De Mita S, Krishnakumar V, Gundlach H, Zhou S, Mudge J, Bharti AK,Murray JD, Naoumkina MA, Rosen B, Silverstein KA, Tang H, Rombauts S, Zhao PX, Zhou P, Barbe V, Bardou P, Bechner M, BellecA, Berger A, Berges H, Bidwell S, Bisseling T, Choisne N, Couloux A, Denny R, Deshpande S, Dai X, Doyle JJ, Dudez AM, FarmerAD, Fouteau S, Franken C, Gibelin C, Gish J, Goldstein S, Gonzalez AJ, Green PJ, Hallab A, Hartog M, Hua A, Humphray SJ, JeongDH, Jing Y, Jocker A, Kenton SM, Kim DJ, Klee K, Lai H, Lang C, Lin S, Macmil SL, Magdelenat G, Matthews L, McCorrison J,Monaghan EL, Mun JH, Najar FZ, Nicholson C, Noirot C, O'Bleness M, Paule CR, Poulain J, Prion F, Qin B, Qu C, Retzel EF, RiddleC, Sallet E, Samain S, Samson N, Sanders I, Saurat O, Scarpelli C, Schiex T, Segurens B, Severin AJ, Sherrier DJ, Shi R, Sims S,Singer SR, Sinharoy S, Sterck L, Viollet A, Wang BB, Wang K, Wang M, Wang X, Warfsmann J, Weissenbach J, White DD, White JD,Wiley GB, Wincker P, Xing Y, Yang L, Yao Z, Ying F, Zhai J, Zhou L, Zuber A, Denarie J, Dixon RA, May GD, Schwartz DC, Rogers J,Quetier F, Town CD, Roe BA (2011) The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480:520-524
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Yu J, Hu S, Wang J, Wong GK-S, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, Cao M, Liu J, Sun J, Tang J, Chen Y, Huang X, Lin W,Ye C, Tong W, Cong L, Geng J, Han Y, Li L, Li W, Hu G, Huang X, Li W, Li J, Liu Z, Li L, Liu J, Qi Q, Liu J, Li L, Li T, Wang X, Lu H,Wu T, Zhu M, Ni P, Han H, Dong W, Ren X, Feng X, Cui P, Li X, Wang H, Xu X, Zhai W, Xu Z, Zhang J, He S, Zhang J, Xu J, Zhang K,Zheng X, Dong J, Zeng W, Tao L, Ye J, Tan J, Ren X, Chen X, He J, Liu D, Tian W, Tian C, Xia H, Bao Q, Li G, Gao H, Cao T, Wang J,Zhao W, Li P, Chen W, Wang X, Zhang Y, Hu J, Wang J, Liu S, Yang J, Zhang G, Xiong Y, Li Z, Mao L, Zhou C, Zhu Z, Chen R, Hao B,Zheng W, Chen S, Guo W, Li G, Liu S, Tao M, Wang J, Zhu L, Yuan L, Yang H (2002) A draft sequence of the rice genome (Oryzasativa L. ssp. indica). Science 296: 79-92
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Zhang G, Liu X, Quan Z, Cheng S, Xu X, Pan S, Xie M, Zeng P, Yue Z, Wang W, Tao Y, Bian C, Han C, Xia Q, Peng X, Cao R, Yang X,Zhan D, Hu J, Zhang Y, Li H, Li H, Li N, Wang J, Wang C, Wang R, Guo T, Cai Y, Liu C, Xiang H, Shi Q, Huang P, Chen Q, Li Y, WangJ, Zhao Z, Wang J (2012) Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuelpotential. Nat Biotechnol 30: 549-554
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Zhang X, Choi J, Heinz J, Chetty C (2006) Domain-specific positive selection contributes to the evolution of Arabidopsis Leucine-Rich Repeat Receptor-Like Kinase (LRR RLK) genes. J Mol Evol 63: 612-621
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Zou C, Lehti-Shiu MD, Thibaud-Nissen F, Prakash T, Buell CR, Shiu S-H (2009) Evolutionary and expression signatures ofpseudogenes in Arabidopsis and rice. Plant Physiol 151: 3-15
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
www.plantphysiol.orgon June 26, 2018 - Published by Downloaded from Copyright © 2016 American Society of Plant Biologists. All rights reserved.