3. operation of the cell at a...
TRANSCRIPT

1
3. Operation of the Cell at a Constant-CurrentThe analytical electrodeposition that we are discussing can be carried out
by maintaining the current, rather than the applied voltage, at a constant level. To maintain constant current, we must increase the applied voltage periodically as the electrolysis proceeds.
It should be emphasized that concentration polarization at the cathode causes a decrease in current. Initially, this effect can be partially offset by increasing the applied potential. Electrostatic forces would then postpone the onset of concentration polarization by enhancing the rate at which, say, copper ions are brought to the electrode surface. Soon, however, the solution becomes so depleted of copper ions that diffusion, electrostatic attraction, and stirring cannot keep the electrode surface supplied with sufficient copper ions to maintain the desired current. When this occurs, further increases in Eappl cause rapid changes in the cathode potential; codeposition of hydrogen (or other reducible species) then takes place.

2
At this point, the cathode potential ultimately becomes stabilized at a level fixed by the standard potential and the overvoltage for the new electrode reaction; further large increases in the cell potential are no longer necessary to maintain a constant current.
Copper continues to deposit as Cu(II) ions reach the electrode surface; the contribution of this process to the total current, however, becomes smaller and smaller as the deposition becomes more and more nearly complete.
The changes in cathode potential under constant current conditions are shown in the following figures:

3
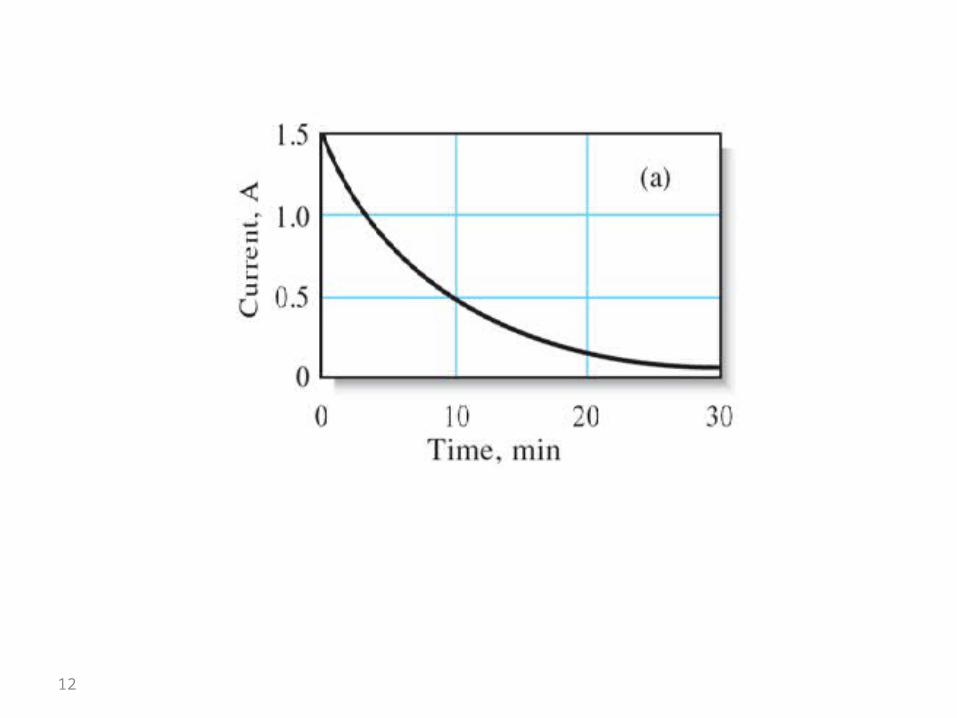
Changes in cathode potential during the deposition of copper with a constant current of 1.5 A. When the potential reaches
that of hydrogen evolution (including overvoltage), the current will be independent on analyte

4

5

6
A constant current of 0.800 A is used to deposit copper at the cathode and oxygen at the anode of an electrolytic cell. Calculate the number of grams of each product formed in 15.2 min, assuming no other redox reaction occurs.
Cu2+ +2e D Cu(s)2H20 D O2(g) + 4H+ + 4e
mol Cu = ½ mol e = ½ * 7.56*10-3 = 3.78*10-3
g Cu = 3.78*10-3 mol Cu* 63.55 (g/mol) = 0.240 gmol O2 = ¼ mol e = ¼ * 7.56*10-3 = 1.89*10-3
g O2 = 1.89*10-3 mol O2* 32.00 (g/mol) = 0.0605 g
𝑚𝑜𝑙 𝑒= 𝐼∗𝑡𝐹 = 0.8 𝐴∗15.2min∗60 ൫𝑠 minൗ� ൯96485 (𝐶 𝑚𝑜𝑙 𝑒ൗ� ) = 729.6 𝐴.𝑠96485 (𝐶 𝑚𝑜𝑙 𝑒ൗ� ) = 7.56∗10−3

7
AN INTRODUCTION TO COULOMETRIC METHODSOF ANALYSIS
Coulometry encompasses a group of analytical methods that involve measuring the quantity of electricity (in coulombs) needed to convert the analyte quantitatively to a different oxidation state. Like gravimetric methods, coulometry has the advantage that calibration or standardization is not usually necessary.
Coulometric methods are often as accurate as gravimetric procedures, and they are usually faster and more convenient.

8
Coulometric as Compared to Other Electroanalytical Methods of Analysis
• Potentiometry: Electrochemical measurements under static current conditions
• Coulometry, electrogravimetry, and voltammetry: Electrochemical measurements under dynamic current conditions (current passes through the cell)
• Coulomteric methods are based on exhaustive electrolysis of the analyte: that is quantitative reduction or oxidation of the analyte at the working electrode or the analyte reacts quantitatively with a reagent generated at the working electrode

9
Units for Quantity of Electricity
The quantity of electricity, or charge, is measured in units of coulombs (C). A coulomb is the quantity of charge transported in one second by a constant current of one ampere. Thus, for a constant current of I amperes for t seconds, the charge in coulombs, Q, is given by the expression: Q = It𝐹= 𝐶𝑚𝑜𝑙 𝑒 , 𝑡ℎ𝑒𝑟𝑒𝑓𝑜𝑟𝑒, 𝑚𝑜𝑙 𝑒= 𝐶𝐹= 𝑄𝐹= 𝐼∗𝑡𝐹
𝑚𝑜𝑙 𝑒= 𝑛∗𝑚𝑜𝑙 𝑎𝑛𝑎𝑙𝑦𝑡𝑒 ሺ𝑛𝐴ሻ

10
The faraday F is the charge in coulombs of one mole of electrons. The charge of the electron is 1.60218*10-19 C, so we may therefore write:
Faraday's law relates the number of moles of the analyte nA to the charge 𝑛𝐴 = 𝑄𝑛𝐹 = 𝐼∗𝑡𝑛𝐹
𝐹= 6.02214∗1023 𝑒𝑚𝑜𝑙 𝑒 ∗ 1.60218∗10−19 𝐶𝑒 = 96485 ቆ𝐶 𝑚𝑜𝑙 𝑒ൗ� ቇ
where n is the number of moles of electrons in the analyte half-reaction.

11
Types of Coulometric MethodsTwo general techniques are used for coulometric analysis:
controlled-potential (potentiostatic) coulometry and controlled-current (amperostatic) coulometry. In controlled-potential coulometry, the potential of the working electrode (the electrode at which the analytical reaction occurs) is maintained at a constant level such that quantitative oxidation or reduction of the analyte occurs without involvement of less-reactive species in the sample or solvent. In this method, the current is initially high but decreases rapidly and approaches zero as the analyte is removed from the solution. The quantity of charge required is usually measured with an electronic integrator.
The end of reaction is identified as the current passing becomes essentially at the value of the background or residual current.
12

13
1 .CONTROLLED-POTENTIAL COULOMETRY
In controlled-potential coulometry, the potential of the working electrode is maintained at a constant level such that the analyte conducts charge across the electrode-solution interface. The charge required to convert the analyte to its reaction product is then determined by integrating the current-versus-time curve during the electrolysis.
An analysis of this kind has all the advantages of an electrogravimetric method, but it is not necessary to weigh a product.
The technique can therefore be applied to systems that yield deposits with poor physical properties as well as to reactions that yield no solid product at all. For example, arsenic may be determined coulometrically by the electrolytic oxidation of arsenous acid (H3AsO3) to arsenic acid (H3AsO4) at a platinum anode.

14
Coulometric methods are performed in much the same way as controlled-potential gravimetric methods.
However, the electrolysis current is recorded as a function of time to give a curve similar to the curve in the side figure. The analysis is then completed by electronically integrating the current-time curve to obtain the number of coulombs.

15
InstrumentationThe instrumentation for potentiostatic coulometry consists of an
electrolysis cell, a potentiostat, and an electronic integrator for determining the charge consumed.
The figure below illustrates two types of cells that are used for potentiostatic coulometry. The first (a) consists of a platinum gauze working electrode and a platinum wire counter electrode, which is separated from the test solution by a porous tube containing the same supporting electrolyte as the test solution. Separating the counter electrode is sometimes necessary to prevent its reaction products from interfering in the analysis. A saturated calomel or a Ag/AgCI reference electrode is connected to the test solution with a salt bridge. Often the bridge contains the same electrolyte as the test solution.

16
The second type of cell (b) is a mercury pool type. A mercury cathode is particularly useful for separating easily reduced elements as a preliminary step in an analysis. For example, copper, nickel, cobalt, silver, and cadmium are readily separated from ions such as aluminum, titanium, the alkali metals, and phosphates. The precipitated elements dissolve in the mercury; little hydrogen evolution occurs even at high applied potentials because of large overvoltage effects. A coulometric cell such as that shown in Figure (b) is also useful for coulometric determination of metal ions and certain types of organic compounds as well.

17

18

19
Potentiostats and Integrators• A three electrode potentiostat system is used. Two types of ‑
working electrodes are commonly used: a Pt electrode manufactured from platinum gauze and fashioned into a ‑cylindrical tube, and an Hg pool electrode.
• The large overpotential for reducing H+ at mercury makes it the electrode of choice for analytes requiring negative potentials. For example, potentials more negative than 1 V ‑versus the SCE are feasible at an Hg electrode (but not at a Pt electrode), even in very acidic solutions.
• The ease with which mercury is oxidized prevents its use at potentials that are positive with respect to the SHE.
• Platinum working electrodes are used when positive potentials are required.

20
• The auxiliary electrode, which is often a Pt wire, is separated by a salt bridge from the solution containing the analyte.
• This is necessary to prevent electrolysis products generated at the auxiliary electrode from reacting with the analyte and interfering in the analysis.
• A saturated calomel or Ag/AgCI electrode serves as the reference electrode.
• A means of determining the total charge passed during electrolysis. One method is to monitor the current as a function of time and determine the area under the curve.
• Modern instruments, however, use electronic integration to monitor charge as a function of time. The total charge can be read directly from a digital readout or from a plot of charge versus time

21
A 0.3619-g of pure tetrachloropicolinic acid, C6HNO2Cl4, is dissolved in distilled water, transferred to a 1000-mL volumetric flask, and diluted to volume. An exhaustive controlled-potential electrolysis of a 10.00-mL portion of this solution at a spongy silver cathode requires 5.374 C of charge. What is the value of n for this reduction reaction?
SolutionThe 10.00-mL portion of sample contains 3.619 mg, or 1.39 × 10–5 mol of
tetrachloropicolinic acid. Solving for n and making appropriate substitutions gives:
n=Q/FNA=5.274C / (96485C/mol e−) * (1.39×10−5 mol C6HNO2Cl4 ) = 3.93 mol e−/mo C6HNO2Cl4
Thus, reducing a molecule of tetrachloropicolinic acid requires four electrons. The overall reaction, which results in the selective formation of 3,6-dichloropicolinic acid, is:

22
2. Controlled-Current Coulometry (Coulometric Titrations)
• The current is kept constant until an indicator signals completion of the analytical reaction.
• The quantity of electricity required to attain the end point is calculated from the magnitude of the current and the time of its passage (Q = I*t).
• Controlled current coulometry, also known as ‑amperostatic coulometry or coulometric titrimetry
• When called coulometric titration, electrons serve as the titrant.

23
Amperostatic versus Potentiostatic Coulometry
Controlled current coulometry, ‑ has two advantages over controlled potential coulometry. ‑
First, using a constant current leads to more rapid analysis since the current does not decrease over time. Thus, a typical analysis time for controlled current coulometry is less than 10 min, as opposed to approximately 30 60 min ‑
for controlled potential coulometry‑ .Second, with a constant current the total charge is simply the product of current and time. A method for integrating the current time curve, therefore, is not necessary‑.

24
Experimental problems with constant current coulometry
Using a constant current does present two important experimental problems that must be solved if accurate results are to be obtained.
First, as electrolysis occurs the analyte's concentration and, therefore, the current due to its oxidation or reduction steadily decreases.
To maintain a constant current the cell potential must change until another oxidation or reduction reaction can occur at the working
electrode .Unless the system is carefully designed, these secondary reactions will
produce a current efficiency of less than 100% .Second problem is the need for a method of determining when the analyte
has been exhaustively electrolyzed (end point). In controlled potential coulometry this is signaled by a decrease in the ‑
current to a constant background or residual current .In controlled current coulometry, a constant current continues to flow ‑even when the analyte has been completely oxidized or reduced. A suitable means of determining the end point of the reaction, t‑ e, is needed.

25
Controlled-current coulometry uses a constant current, which passes through the cell until an indicator signals completion of the analytical reaction. The quantity of charge required to reach the end point is then calculated from the magnitude of the current and the time that the current passes. This method has enjoyed wider application than potentiostatic coulometry. It is frequently called a coulometric titration. A fundamental requirement of all coulometric methods is that the analyte must react with 100% current efficiency. This requirement means that each faraday of charge must bring about a chemical change in (1/n) of the moles of analyte, where n is the number of electrons that is equivalent to one mole of analyte.𝐹= 𝐶𝑚𝑜𝑙 𝑒 , 𝑚𝑜𝑙 𝑒= 𝑛∗𝑚𝑜𝑙 𝑎𝑛𝑎𝑙𝑦𝑡𝑒 ሺ𝑛𝐴ሻ
𝐹= 𝐶𝑛∗𝑚𝑜𝑙 𝑎𝑛𝑎𝑙𝑦𝑡𝑒 ሺ𝑛𝐴ሻ

26
Current efficiency of 100% does not, however, imply that the analyte must necessarily participate directly in the electron-transfer process at the electrode. Indeed, more often than not, the analyte participates, at least in part, in a reaction that is secondary to the electrode reaction. For example, at the beginning of the oxidation of iron(II) at the anode, all of the current results from the reaction:
Fe2+ D Fe3+ + eAs the concentration of iron(II) decreases, however,
concentration polarization will cause the anode potential to rise until decomposition (oxidation) of water occurs as a competing process. That is:
2H20 D O2(g) + 4H+ + 4e

27
The charge required to completely oxidize all of the iron(Il) in the solution would then exceed that demanded by theory (as it will belong to oxidizing both iron and water). To avoid the resulting error, an unmeasured excess of cerium(III) can be introduced at the start of the electrolysis. This ion is oxidized at a lower anode potential than water:
Ce3+ D Ce4+ + e The cerium(IV) produced diffuses rapidly from the electrode
surface, where it then oxidizes an equivalent amount of iron(II):Fe2+ + Ce4+ D Fe3+ + Ce3+ The net effect is an electrochemical oxidation of iron(II) with 100%
current efficiency even though only a fraction of the iron(II) ions are directly oxidized at the electrode surface.
An indicator system should be used to tell at the first appearance of Ce4+ in solution.

28
Maintaining Current Efficiency
Why changing the working electrode's potential can lead to less than 100% current efficiency?
let's consider the coulometric analysis of Fe2+ based on its oxidation to Fe3+ at the working electrode.
Fe2+(aq) D Fe3+(aq) + e ‑
Initially the potential of the working electrode remains nearly constant at a level near the standard electrode potential of the Fe3+/Fe2+ redox couple.
As the concentration of Fe2+ decreases, the potential of the working electrode shifts toward more positive values until another oxidation reaction can provide the necessary current.
Thus, in this case the potential eventually increases to a level at which the oxidation of H2O occurs.
2H20 D O2(g) + 4H+ + 4e

29
• Since the current due to the oxidation of H2O does not contribute to the oxidation of Fe2+, the current efficiency of the analysis is less than 100%.
• To maintain a 100% current efficiency, the products of any competing oxidation reactions must react both rapidly and quantitatively with the remaining Fe2+.
• This may be accomplished, for example, by adding an excess of Ce3+ to the analytical solution.
• When the potential of the working electrode shifts to a more positive potential, the first species to be oxidized is Ce3+.
• Ce3+(aq) D Ce4+(aq) + e‑
• The Ce4+ produced at the working electrode rapidly mixes with the solution, where it reacts with any available Fe2+.
• The first persistent appearance of Ce4+ indicates the end point.

30
Ce4+(aq) + Fe2+(aq) D Fe 3+(aq) + Ce3+(aq)Combining these reactions gives the desired overall reaction Fe 2+(aq) D Fe3+(aq) + e-
Thus, a current efficiency of 100% is maintained. Since the concentration of Ce3+ remains at its initial level, the
potential of the working electrode remains constant as long as any Fe 2+ is present.
This prevents other oxidation reactions, such as that for oxidation of H2O, from interfering with the analysis.
A species, such as Ce3+ which is used to maintain 100% current efficiency is called a Mediator.

31
The coulometric determination of chloride provides another example of an indirect process. Here, a silver electrode is the anode, and silver ions are produced by the current. These cations diffuse into the solution and precipitate the chloride. A current efficiency of 100% with respect to the chloride ion is achieved even though this ion is neither oxidized nor reduced in the cell.
The first persistent appearance of Ag+ indicates the end point.

32
A Typical Coulometric Titration Cell

Coulometric titration of hydrochloric acid
Many chemical methods of analysis are based on reactions among the analyte and a titration reagent that is added to the sample in a measurable form. For example, in a volumetric method (titration), the reagent is added in the form of a solution with an accurately known concentration. The amount of analyte is finally calculated by means of the volume of reagent solution that was necessary to convert the analyte into a product and the stoichiometric equation is used for the calculation.

In a coulometric titration, the reagent is generated by an electrochemical reaction that occurs in the sample itself. An electrochemical cell is built-up; it contains the sample solution (with some suitable additives) as electrolyte, and a pair of electrodes that convey electricity to the system. In addition, the experimental set-up includes a system for monitoring the progress of the reaction in order to detect the end point. Let us look at a sample of HCl that is titrated by OH ions produced by electrolysis at the Pt cathode according to the following reactions:
(1) 2 H2O + 2 e- g 2 OH-(aq) + H2(g) (2) H+(sample) + OH-g H2O

Due to the above reactions, solution pH varies and allows monitoring the progress of the titration using a pH meter. The anode consists of a silver wire and the anode reaction occurs as follows:
(3) Ag(s) g Ag+(aq) + e-
A halogen ion (e.g. Br-) should be added to the sample in order to form a sparingly soluble compound that prevents Ag+ from reaching the cathode:
(4) Ag+(aq) + Br-(aq) g AgBr(s) If no Br- is present in the solution, Ag+ can reach the cathode
and get reduced (according to the reverse of reaction No. 3). This reaction would compete with OH- production (reaction 1) that is the single cathode reaction expected to occur.

Dissolved oxygen should be removed from the sample by a stream of pure nitrogen. Otherwise, oxygen undergoes a cathode reaction (No. 5) which consumes electrons in addition to reaction No. (1) and leads to a positive error.
(5) O2(aq) + 4e- + 4H+ g 2H2O The nitrogen stream also removes dissolved carbon dioxide
which imparts the solution some acidity. The titration proceeds at a constant electric current (constant current coulometry). OH- ions (that are produced according to reaction (1)) diffuse rapidly into the solution and reacts with H+ ions in the sample.

The amount of HCl (nHCl, in mole) is the same as the amount of OH- produced by electrolysis. The number of moles of OH- (nOH) generated (reacted) can be calculated from reaction (1):
nOH = I*t/nFHere, I is the current (in A), t is the electrolysis time (in
seconds), n is the number of electrons per OH- ion in the cathode reaction (1), and F is the Faraday constant (in coulombs per mole of electrons).
In order to monitor the progress of the titration, pH is recorded and plotted as a function of electrolysis time (see figure below). The end point is indicated by an abrupt increase in pH and the time corresponding to pH = 7 (te) indicates the end of the neutralization process.
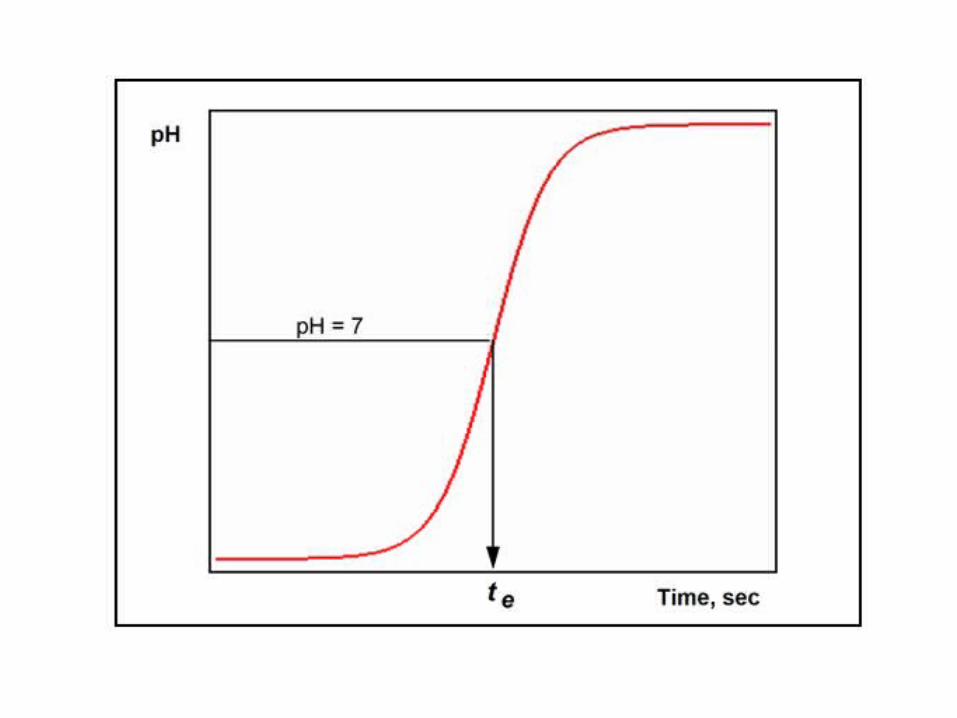

The amount of HCl in the sample (in moles) is:nHCl = I*te/nFClearly, the accuracy of the final result depends on the
accuracy in te determination.The experimental setup is as described previously. The
cathode (-) should be in the form of a Pt plate, spiral or mesh, in order to secure a large enough surface area. The anode (+) is a silver spiral or rod. The pH meter, which is used to monitor pH variation during the titration, should be calibrated prior to the titration by means of suitable pH standards. Phenolphthalein can also be use to detect the end-point by color change.

40
End point detection in coulometric titrations
Coulometric titrations require a means for determining when reaction between analyte and reagent is complete. End point can be detected by visual methods (by using coloured acid-base and redox indicators).
Instrumental techniques of end point detection:1. potentiometric methods2. amperometric methods3. spectrophotometric methods

41
Conceptual diagram of a coulometric titration apparatus. Commercial coulometric titrators are totally electronic and usually computer controlled.

42
Advantages of coulometric titration versus volumetric titration :
1. No need to prepare, standardize, or store standard solutions.
2. Can prepare unstable reagents in situ, since they react almost as soon as they are generated – e.g., Cl2, Br2
3. Straightforward to generate tiny quantities of reagent with good accuracy since it is easy to control current and time electronically.
4. A single coulometric titration apparatus can be used for redox, acid/base, precipitation, complexometric, etc., titrations

43
Examples
To determine the purity of a sample of Na2S2O3, a sample is titrated coulometrically using I– as a mediator and I3
– as the internally generated titrant. A sample weighing 0.1342 g is transferred to a 100-mL volumetric flask and diluted to volume with distilled water. A 10.00-mL portion is transferred to an electrochemical cell along with 25 mL of 1 M KI, 75 mL of a pH 7.0 phosphate buffer, and several drops of a starch indicator solution. Electrolysis at a constant current of 36.45 mA requires 221.8 s to reach the starch indicator endpoint. Determine the sample’s purity.

44
Explanation:At the onset of titration, S2O3
2-(aq) is oxidized to S4O62- at
the anode, then with time the concentration of S2O32-
decreases significantly and thus the potential increases. As the potential reaches that of the mediator redox
potential, the mediator (I−) is oxidized to I3− . The reaction
then becomes that of the generated I3− with S2O3
2- till the end point of the titration, where all S2O3
2- is consumed, and the solution becomes blue as I3
− reacts with starch at the very first appearance of excess I3
−.

45
Solution2S2O3
2-(aq)+ I3− (aq) S⇋ 4O6
2- (aq)+3I−(aq)The oxidation of S2O3
2– to S4O62– requires one electron per S2O3
2– (n = 1).
solving for the moles and grams of Na2S2O3 gives:NA=I*t/nF=(0.03645A)(221.8s)/96485 = 8.379×10−5 mol Na2S2O3
g Na2S2O3 = 8.379×10−5 mol Na2S2O3 *158 g Na2S2O3 /mol = 0.01325g
This is the amount of Na2S2O3 in a 10.00-mL portion of a 100-mL sample; thus, there are 0.1325 grams of Na2S2O3 in the original sample. The sample’s purity, therefore, is 98.73%
Note that, it does not matter whether S2O32– is oxidized directly
at the working electrode or is indirectly oxidized by I3–.

46
A 2.00 mL volume contains 0.6113 mg/mL cyclohexene. How much time is required at constant current of 4.825 mA to convert cyclohexene to the dibromo derivative?
C6H10 + Br2 g C6H10Br2
Br2 + 2e g 2 Br-
mol e = 2*mol cyclohexenemol e = I*t/F or t = {mol e * F/I}
𝑡 = ൫2∗1.49∗10−5𝑚𝑜𝑙 𝑒൯∗(96485 𝐶 𝑚𝑜𝑙 𝑒ൗ� )(4.825∗10−3 𝐶 𝑆ൗ�) = 595 𝑠
𝑔 𝑐𝑦𝑐𝑙𝑜ℎ𝑒𝑥𝑒𝑛𝑒= 𝑔𝑚𝐿∗𝑉𝑚𝐿=0.6113∗10−3𝑚𝐿 ∗2.00 𝑚𝐿= 1.2226∗ 10−3 𝑚𝑜𝑙 𝑐𝑦𝑐𝑙𝑜ℎ𝑒𝑥𝑒𝑛𝑒= 𝑔𝐹𝑊 = 1.2226∗ 10−382.15 =1.49∗10−5