2016-02-04 whole genome data for the analysis of food ... · whole genome data for the analysis of...
TRANSCRIPT

Whole genome data for the analysisof food borne infections
Martin Maiden
NIHR HPRU in Gastrointestinal InfectionsNIHR HPRU in Gastrointestinal Infections
Department of Zoology, University of Oxford

Acknowledgements
Clare Barker
Julia Bennett
Carly Bliss
Holly Bratcher
James Bray
Dorothea Hill
Lisa Rebbets
Melissa Jansen vanRensburg
Keith JolleyJames Bray
Carina Brehony
Marianne Clemence
Ali Cody
Fran Colles
Kanny DialloWTF
Sarah Earle
Suzanne Ford
Odile HarrisonWT
Sofia Hauck
Keith Jolley
Jasna Kovac
Jenny MacLennan
Noel McCarthy
Maddi Pearce
Charlene Rodrigues
Samuel Sheppard
Helen Strain
Eleanor Watkins
Helen Wimalarathna

Acknowledgements

Genome sequences and clinicalmicrobiology
• Definitive:
– all biological variation ultimately derives fromnucleotide sequence changes;
– fundamental level of information;
– any part of the genome can be accessed.
• Reproducible:
– nucleotide sequences are either right or wrong
1 gagttttatc gcttccatga cgcagaagtt aacactttcg gatatttctg atgagtcgaa61 aaattatctt gataaagcag gaattactac tgcttgttta cgaattaaat cgaagtggac121 tgctggcgga aaatgagaaa attcgaccta tccttgcgca gctcgagaag ctcttacttt181 gcgacctttc gccatcaact aacgattctg tcaaaaactg acgcgttgga tgaggagaag241 tggcttaata tgcttggcac gttcgtcaag gactggttta gatatgagtc acattttgtt301 catggtagag attctcttgt tgacatttta aaagagcgtg gattactatc tgagtccgat361 gctgttcaac cactaatagg taagaaatca tgagtcaagt tactgaacaa tccgtacgtt421 tccagaccgc tttggcctct attaagctca ttcaggcttc tgccgttttg gatttaaccg481 aagatgattt cgattttctg acgagtaaca aagtttggat tgctactgac cgctctcgtg541 ctcgtcgctg cgttgaggct tgcgtttatg gtacgctgga ctttgtggga taccctcgct601 ttcctgctcc tgttgagttt attgctgccg tcattgctta ttatgttcat cccgtcaaca661 ttcaaacggc ctgtctcatc atggaaggcg ctgaatttac ggaaaacatt attaatggcg721 tcgagcgtcc ggttaaagcc gctgaattgt tcgcgtttac cttgcgtgta cgcgcaggaa781 acactgacgt tcttactgac gcagaagaaa acgtgcgtca aaaattacgt gcggaaggag841 tgatgtaatg tctaaaggta aaaaacgttc tggcgctcgc cctggtcgtc cgcagccgtt901 gcgaggtact aaaggcaagc gtaaaggcgc tcgtctttgg tatgtaggtg gtcaacaatt961 ttaattgcag gggcttcggc cccttacttg aggataaatt atgtctaata ttcaaactgg
1021 cgccgagcgt atgccgcatg acctttccca tcttggcttc cttgctggtc agattggtcg1081 tcttattacc atttcaacta ctccggttat cgctggcgac tccttcgaga tggacgccgt1141 tggcgctctc cgtctttctc cattgcgtcg tggccttgct attgactcta ctgtagacat1201 ttttactttt tatgtccctc atcgtcacgt ttatggtgaa cagtggatta agttcatgaa1261 ggatggtgtt aatgccactc ctctcccgac tgttaacact actggttata ttgaccatgc1321 cgcttttctt ggcacgatta accctgatac caataaaatc cctaagcatt tgtttcaggg1381 ttatttgaat atctataaca actattttaa agcgccgtgg atgcctgacc gtaccgaggc1441 taaccctaat gagcttaatc aagatgatgc tcgttatggt ttccgttgct gccatctcaa1501 aaacatttgg actgctccgc ttcctcctga gactgagctt tctcgccaaa tgacgacttc1561 taccacatct attgacatta tgggtctgca agctgcttat gctaatttgc atactgacca1621 agaacgtgat tacttcatgc agcgttacca tgatgttatt tcttcatttg gaggtaaaac1681 ctcttatgac gctgacaacc gtcctttact tgtcatgcgc tctaatctct gggcatctgg1741 ctatgatgtt gatggaactg accaaacgtc gttaggccag ttttctggtc gtgttcaaca1801 gacctataaa cattctgtgc cgcgtttctt tgttcctgag catggcacta tgtttactct1861 tgcgcttgtt cgttttccgc ctactgcgac taaagagatt cagtacctta acgctaaagg1921 tgctttgact tataccgata ttgctggcga ccctgttttg tatggcaact tgccgccgcg1981 tgaaatttct atgaaggatg ttttccgttc tggtgattcg tctaagaagt ttaagattgc2041 tgagggtcag tggtatcgtt atgcgccttc gtatgtttct cctgcttatc accttcttga2101 aggcttccca ttcattcagg aaccgccttc tggtgatttg caagaacgcg tacttattcg2161 ccaccatgat tatgaccagt gtttccagtc cgttcagttg ttgcagtgga atagtcaggt
– nucleotide sequences are either right or wrongand this can be checked;
– reverse mutations are (usually) rare.
• Scalable:
– nucleotide sequencing technology can beconducted on one or many samples and on a fewbase pairs or a whole genome.
• Manipulable:
– nucleotide sequences;
– can be analysed with model-based methods.
2221 taaatttaat gtgaccgttt atcgcaatct gccgaccact cgcgattcaa tcatgacttc2281 gtgataaaag attgagtgtg aggttataac gccgaagcgg taaaaatttt aatttttgcc2341 gctgaggggt tgaccaagcg aagcgcggta ggttttctgc ttaggagttt aatcatgttt2401 cagactttta tttctcgcca taattcaaac tttttttctg ataagctggt tctcacttct2461 gttactccag cttcttcggc acctgtttta cagacaccta aagctacatc gtcaacgtta2521 tattttgata gtttgacggt taatgctggt aatggtggtt ttcttcattg cattcagatg2581 gatacatctg tcaacgccgc taatcaggtt gtttctgttg gtgctgatat tgcttttgat2641 gccgacccta aattttttgc ctgtttggtt cgctttgagt cttcttcggt tccgactacc2701 ctcccgactg cctatgatgt ttatcctttg aatggtcgcc atgatggtgg ttattatacc2761 gtcaaggact gtgtgactat tgacgtcctt ccccgtacgc cgggcaataa cgtttatgtt2821 ggtttcatgg tttggtctaa ctttaccgct actaaatgcc gcggattggt ttcgctgaat2881 caggttatta aagagattat ttgtctccag ccacttaagt gaggtgattt atgtttggtg2941 ctattgctgg cggtattgct tctgctcttg ctggtggcgc catgtctaaa ttgtttggag3001 gcggtcaaaa agccgcctcc ggtggcattc aaggtgatgt gcttgctacc gataacaata3061 ctgtaggcat gggtgatgct ggtattaaat ctgccattca aggctctaat gttcctaacc3121 ctgatgaggc cgcccctagt tttgtttctg gtgctatggc taaagctggt aaaggacttc3181 ttgaaggtac gttgcaggct ggcacttctg ccgtttctga taagttgctt gatttggttg3241 gacttggtgg caagtctgcc gctgataaag gaaaggatac tcgtgattat cttgctgctg3301 catttcctga gcttaatgct tgggagcgtg ctggtgctga tgcttcctct gctggtatgg3361 ttgacgccgg atttgagaat caaaaagagc ttactaaaat gcaactggac aatcagaaag3421 agattgccga gatgcaaaat gagactcaaa aagagattgc tggcattcag tcggcgactt3481 cacgccagaa tacgaaagac caggtatatg cacaaaatga gatgcttgct tatcaacaga3541 aggagtctac tgctcgcgtt gcgtctatta tggaaaacac caatctttcc aagcaacagc3601 aggtttccga gattatgcgc caaatgctta ctcaagctca aacggctggt cagtatttta3661 ccaatgacca aatcaaagaa atgactcgca aggttagtgc tgaggttgac ttagttcatc3721 agcaaacgca gaatcagcgg tatggctctt ctcatattgg cgctactgca aaggatattt3781 ctaatgtcgt cactgatgct gcttctggtg tggttgatat ttttcatggt attgataaag3841 ctgttgccga tacttggaac aatttctgga aagacggtaa agctgatggt attggctcta3901 atttgtctag gaaataaccg tcaggattga caccctccca attgtatgtt ttcatgcctc3961 caaatcttgg aggctttttt atggttcgtt cttattaccc ttctgaatgt cacgctgatt4021 attttgactt tgagcgtatc gaggctctta aacctgctat tgaggcttgt ggcatttcta4081 ctctttctca atccccaatg cttggcttcc ataagcagat ggataaccgc atcaagctct4141 tggaagagat tctgtctttt cgtatgcagg gcgttgagtt cgataatggt gatatgtatg4201 ttgacggcca taaggctgct tctgacgttc gtgatgagtt tgtatctgtt actgagaagt4261 taatggatga attggcacaa tgctacaatg tgctccccca acttgatatt aataacacta4321 tagaccaccg ccccgaaggg gacgaaaaat ggtttttaga gaacgagaag acggttacgc4381 agttttgccg caagctggct gctgaacgcc ctcttaagga tattcgcgat gagtataatt4441 accccaaaaa gaaaggtatt aaggatgagt gttcaagatt gctggaggcc tccactatga4501 aatcgcgtag aggctttgct attcagcgtt tgatgaatgc aatgcgacag gctcatgctg4561 atggttggtt tatcgttttt gacactctca cgttggctga cgaccgatta gaggcgtttt4621 atgataatcc caatgctttg cgtgactatt ttcgtgatat tggtcgtatg gttcttgctg4681 ccgagggtcg caaggctaat gattcacacg ccgactgcta tcagtatttt tgtgtgcctg4741 agtatggtac agctaatggc cgtcttcatt tccatgcggt gcactttatg cggacacttc4801 ctacaggtag cgttgaccct aattttggtc gtcgggtacg caatcgccgc cagttaaata4861 gcttgcaaaa tacgtggcct tatggttaca gtatgcccat cgcagttcgc tacacgcagg4921 acgctttttc acgttctggt tggttgtggc ctgttgatgc taaaggtgag ccgcttaaag4981 ctaccagtta tatggctgtt ggtttctatg tggctaaata cgttaacaaa aagtcagata5041 tggaccttgc tgctaaaggt ctaggagcta aagaatggaa caactcacta aaaaccaagc5101 tgtcgctact tcccaagaag ctgttcagaa tcagaatgag ccgcaacttc gggatgaaaa5161 tgctcacaat gacaaatctg tccacggagt gcttaatcca acttaccaag ctgggttacg5221 acgcgacgcc gttcaaccag atattgaagc agaacgcaaa aagagagatg agattgaggc5281 tgggaaaagt tactgtagcc gacgttttgg cggcgcaacc tgtgacgaca aatctgctca5341 aatttatgcg cgcttcgata aaaatgattg gcgtatccaa cctgca
Sanger, F., Air, G. M., Barrell, B. G., Brown, N. L.,Coulson, A. R., Fiddes, C. A., Hutchison, C. A.,Slocombe, P. M. & Smith, M. (1977). Nucleotidesequence of bacteriophage phi X174 DNA. Nature265, 687-695.
Frederick Sanger (1918-2013)

Questions in clinical microbiology
Centuries+ decades years months weeks days hours
evolution emergence epidemiology diagnosis
Relative discrimination required HighLow
Relative amount of genetic change LowHigh
Mice andmen
Vibrio cholerae
Yersinia pestis
Escherichia coli
Plants
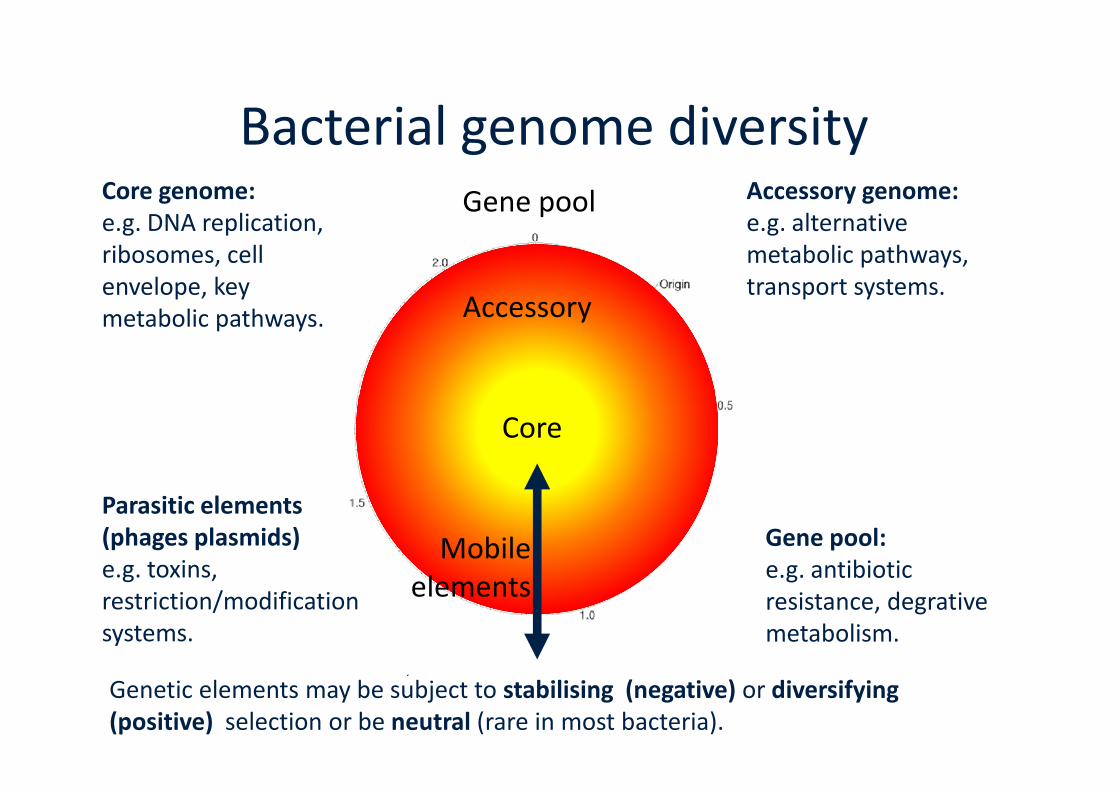
Bacterial genome diversityGene pool
Accessory
Accessory genome:e.g. alternativemetabolic pathways,transport systems.
Core genome:e.g. DNA replication,ribosomes, cellenvelope, keymetabolic pathways.
Mycoplamagenitalium
Streptococcuspneumoniae
Bacillusanthracis
Mycobacteriumtuberculosis
Campylobacterjejuni
Neisseriameningitidis
TreponemapallidumChlamydia
pneumoniae
Rickettsiaprowazekii
Staphylococcusaureus
Genetic elements may be subject to stabilising (negative) or diversifying(positive) selection or be neutral (rare in most bacteria).
Parasitic elements(phages plasmids)e.g. toxins,restriction/modificationsystems.
Core
Mobileelements
Gene pool:e.g. antibioticresistance, degrativemetabolism.

Patterns in sequence variationa primer on bacterial population biology
Ideas about bacterialpopulations are dominated thefacts that bacteria:• are asexual;• reproduce by binary fission,
with each ‘mother’ cellwith each ‘mother’ cellgiving rise to two identical‘daughter’ cells (clones).
• accumulate genetic changeby ‘vertical’ inheritance. Gupta, S. & Maiden, M.C.J. Exploring
the evolution of diversity in pathogenpopulations. Trends in Microbiology 9,181-192 (2001).

The clonal population model:asexuality with diversity reduction
Diversity reduction
periodicselection
Original genotype
Mutant genotype
selection
bottlenecking
Levin BR. 1981. Periodicselection, infectious geneexchange and the geneticstructure of E. coli populations.Genetics 99(1):1-23.
Bacterial populations should, therefore, be easy to understand …

Molecular typing made easy – theclonal frame
Years
100s+
decades years weeks/
months
hours/
days
AAAA
ATAT
ATAT
Progressive accumulation of genetic change
AAAA
ATAA
ATAT
ATAA
TTAT
TTTT
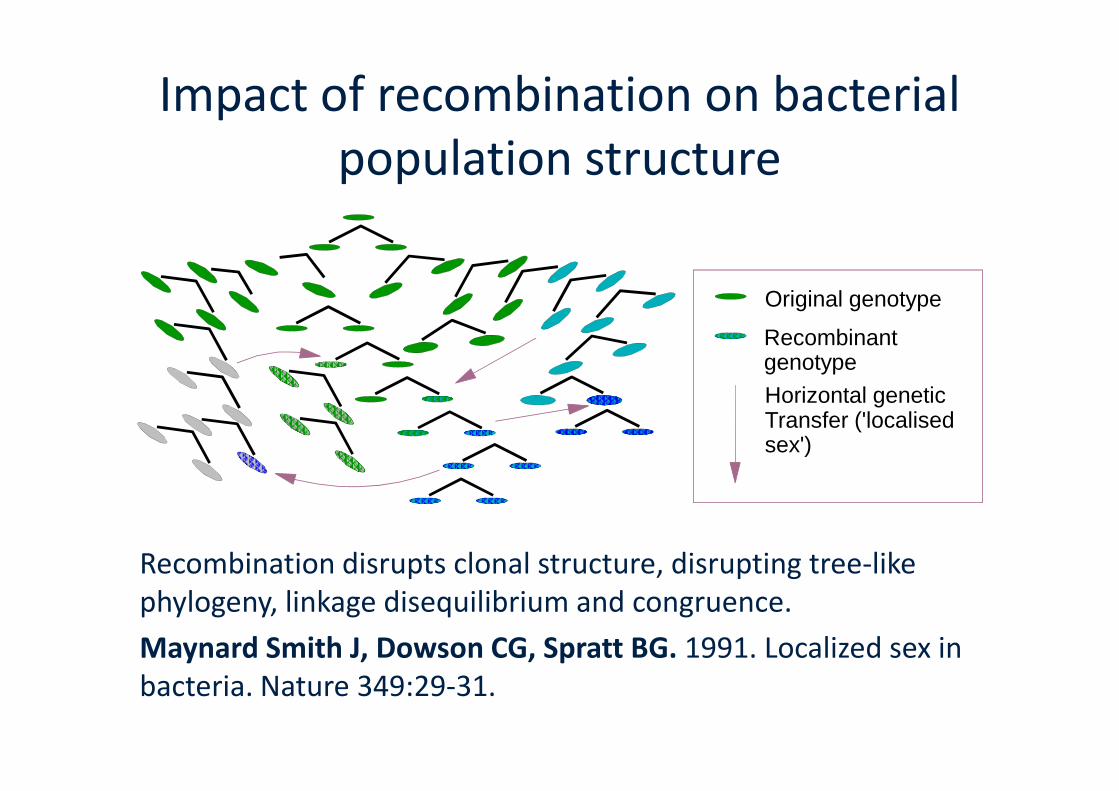
Impact of recombination on bacterialpopulation structure
Horizontal genetic
Original genotype
Recombinantgenotype
Recombination disrupts clonal structure, disrupting tree-likephylogeny, linkage disequilibrium and congruence.
Maynard Smith J, Dowson CG, Spratt BG. 1991. Localized sex inbacteria. Nature 349:29-31.
Horizontal geneticTransfer ('localisedsex')

Clonal and non-clonal populationstructures
Clonal
• Linkage disequilibrium
– non-random allelecombinations.
• Tree-like phylogeny
Non-clonal
• Linkage equilibrium
– random allelecombinations.
• Net-like phylogeny• Tree-like phylogeny
– a bifurcating tree accuratelymodels descent.
• Congruence
– the same phylogentic signalis recorded throughout thegenome
• Net-like phylogeny
– a bifurcating treecannot model descent.
• Incongruence
– different phylogenticsignals are recordedthroughout the genome

A spectrum of population structuresStrictly clonal Fully non-clonal
In practice, different levels of clonal signal is observed in
C. jejuni H. pyloriS. entericaS. Typhi
In practice, different levels of clonal signal is observed indifferent bacterial populations.It is thought that this is a consequence of differingrelative rates of recombination to mutation, althoughother forces may play a role.
Gupta, S. & Maiden, M.C.J. Exploring the evolution ofdiversity in pathogen populations. Trends in Microbiology9, 181-192 (2001).

Dealing with recombination
• Recombination violates the assumptions of clonalevolution.
• This has to be accounted for in models ofbacterial evolution by:– Ignoring it (works if recombination is very low);– Ignoring it (works if recombination is very low);– Identifying (always an estimation) and
• removing possible recombination (e.g. GUBBINS), or• Including recombination and mutation in calculation of
phylogenies (CLONALFRAME and CLONALFRAMEML);
– Using alleles as the unit of analysis (gene-by-geneanalysis).
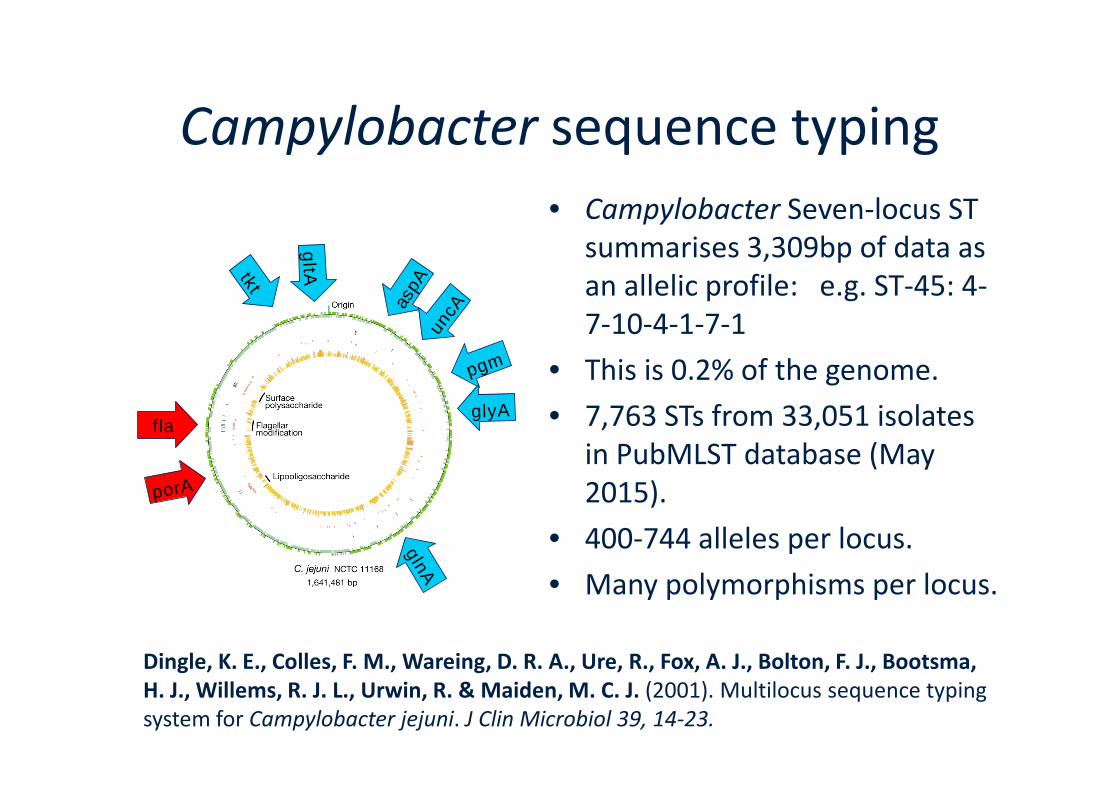
Campylobacter sequence typing
tkt
gltA
aspA
uncA
pgm
10 20 30 40 50aspA1 ATGATAGGTGAAGATATACAAAGAGTATTAGAAGCTAGAAAATTGATTTTaspA2 ..................................................aspA3 ..................................................aspA4 ........C...................................A.....aspA6 ..................................................aspA7 ..................................................aspA8 ..................................................aspA9 ..................................................aspA10 ........C...................................A.....aspA14 ..................................................aspA16 ..................................................aspA17 ..................................................
• Campylobacter Seven-locus STsummarises 3,309bp of data asan allelic profile: e.g. ST-45: 4-7-10-4-1-7-1
• This is 0.2% of the genome.
Dingle, K. E., Colles, F. M., Wareing, D. R. A., Ure, R., Fox, A. J., Bolton, F. J., Bootsma,H. J., Willems, R. J. L., Urwin, R. & Maiden, M. C. J. (2001). Multilocus sequence typingsystem for Campylobacter jejuni. J Clin Microbiol 39, 14-23.
glyA
gln
A
porA
fla
aspA18 ..................................................aspA19 ..................................................aspA20 ........C.........................................aspA21 ..................................................aspA22 ..................................................aspA23 ..................................................aspA24 ..................................................aspA26 ..................................................aspA27 ..................................................aspA28 .................G................................aspA30 ............................................A.....aspA31 ..................................................aspA32 .....G....................T...............C.......aspA33 .....G....................T...............C.......aspA34 ..................................................aspA48 ........C...................................A.....aspA64 ..................................................
• 7,763 STs from 33,051 isolatesin PubMLST database (May2015).
• 400-744 alleles per locus.
• Many polymorphisms per locus.

Polymorphisms in the uncA MLST locus

Relationships among STs:clonal complexes
ST-45, 26 isolates
ST-4917, 1 isolate4-280-10-4-1-7-1
ST-1701, 1 isolate4-7-10-4-1-51-1
glnA
tkt
3nt
1 ntST-45, 26 isolates4-7-10-4-1-7-1
4-7-10-4-1-51-1
ST-137, 9 isolates4-7-10-4-42-7-1
ST-583, 4 isolates4-7-10-4-42-51-1
ST-2219, 2 isolates10-7-10-4-1-7-1
ST-4852, 1 isolate37-7-10-4-1-7-1
ST-5086, 1 isolate7-7-10-4-1-7-1
tkt
tkt
pgm pgm
aspA
Colles, F. M. & Maiden, M. C. J. (2012). Campylobacter sequence typing:applications and future prospects. Microbiology. 158(11): 2695-2709.
2 nt
7 nt
9 nt
1 nt
60
70
80
90
100
Pe
rce
nt
of
Iso
late
s
humans
poultry
cattle
sheep
pigs
starlings
sand
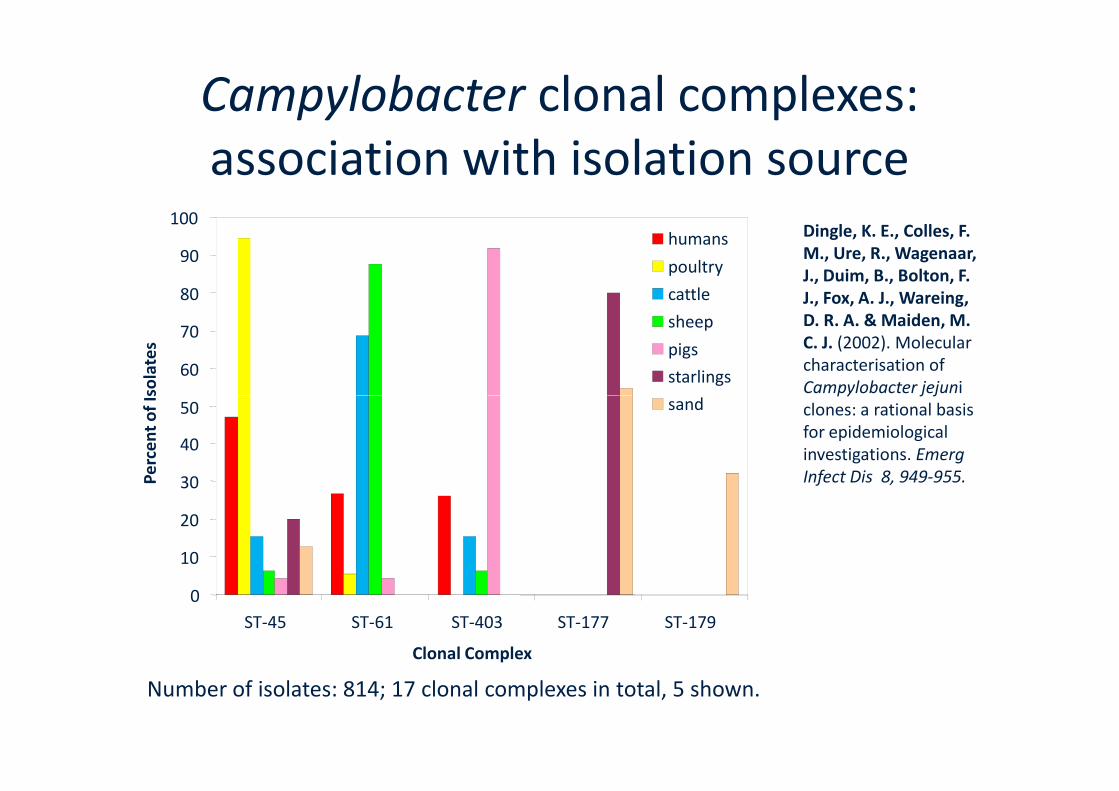
Dingle, K. E., Colles, F.M., Ure, R., Wagenaar,J., Duim, B., Bolton, F.J., Fox, A. J., Wareing,D. R. A. & Maiden, M.C. J. (2002). Molecularcharacterisation ofCampylobacter jejuni
Campylobacter clonal complexes:association with isolation source
0
10
20
30
40
50
ST-45 ST-61 ST-403 ST-177 ST-179
Clonal Complex
Pe
rce
nt
of
Iso
late
s
sand
Number of isolates: 814; 17 clonal complexes in total, 5 shown.
Campylobacter jejuniclones: a rational basisfor epidemiologicalinvestigations. EmergInfect Dis 8, 949-955.
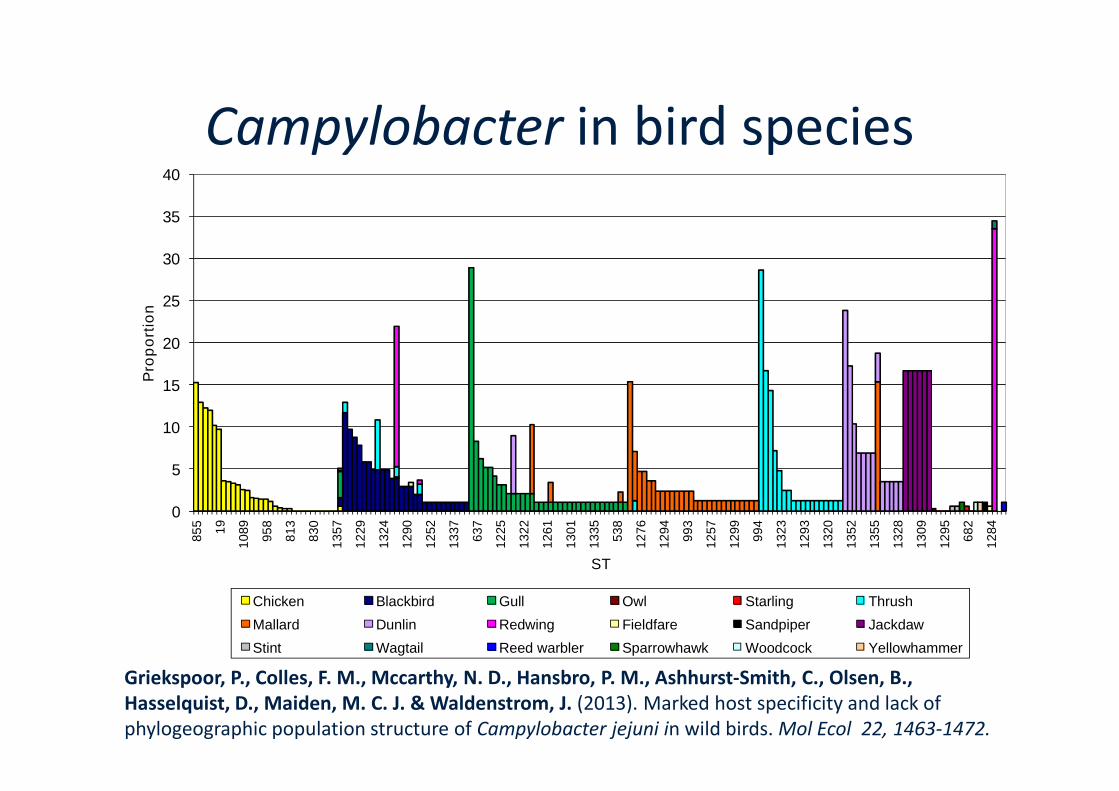
15
20
25
30
35
40
Pro
po
rtio
nCampylobacter in bird species
0
5
10
85
5
19
10
89
95
8
81
3
83
0
13
57
12
29
13
24
12
90
12
52
13
37
63
7
12
25
13
22
12
61
13
01
13
35
53
8
12
76
12
94
99
3
12
57
12
99
99
4
13
23
12
93
13
20
13
52
13
55
13
28
13
09
12
95
68
2
12
84
ST
Chicken Blackbird Gull Owl Starling Thrush
Mallard Dunlin Redwing Fieldfare Sandpiper Jackdaw
Stint Wagtail Reed warbler Sparrowhawk Woodcock Yellowhammer
Griekspoor, P., Colles, F. M., Mccarthy, N. D., Hansbro, P. M., Ashhurst-Smith, C., Olsen, B.,Hasselquist, D., Maiden, M. C. J. & Waldenstrom, J. (2013). Marked host specificity and lack ofphylogeographic population structure of Campylobacter jejuni in wild birds. Mol Ecol 22, 1463-1472.

Campylobacter Genealogies and clonalcomplexes
21 28345
22
48
353
682
443
354
ST-21 Complex
ST-257Complex
ST-45Complex
ST-42Complex
0.02
0.02
177
403
179
52
42
C. doylei
1275206
433
257
61
443
ST-61Complex
Bovine/ovine
Environment
Human disease
Chicken
Both
Sheppard, S. K., Colles, F. M., McCarthy, N. D., Strachan, N. J., Ogden, I. D., Forbes, K.J., Dallas, J. F. & Maiden, M. C. (2011). Niche segregation and genetic structure ofCampylobacter jejuni populations from wild and agricultural host species. Mol Ecol 20,3484-3490.
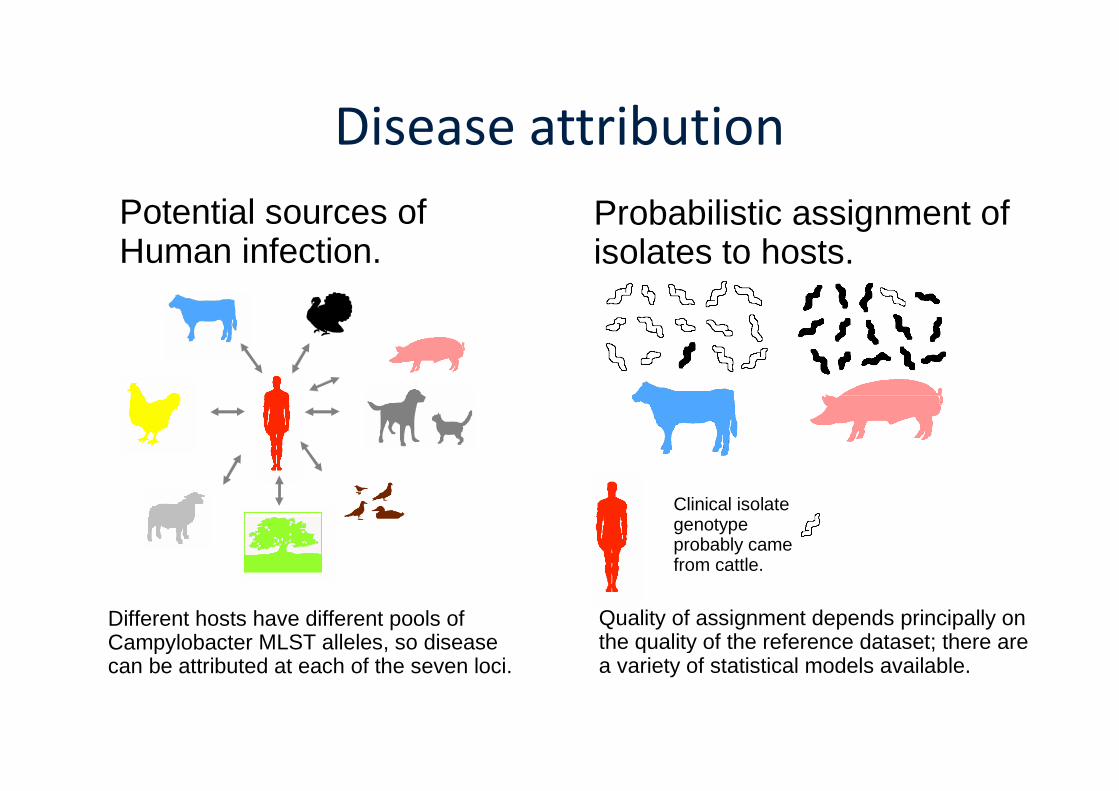
Disease attribution
Potential sources ofHuman infection.
Probabilistic assignment ofisolates to hosts.
Different hosts have different pools ofCampylobacter MLST alleles, so diseasecan be attributed at each of the seven loci.
Clinical isolategenotypeprobably camefrom cattle.
Quality of assignment depends principally onthe quality of the reference dataset; there area variety of statistical models available.

Attribution of Campylobacter tochicken, UK & NZ
Wilson, D. J., Gabriel, E., Leatherbarrow, A. J. H., Cheesbrough, J., Gee, S., Bolton, E., Fox, A., Fearnhead, P., Hart, A. & Diggle, P. J. (2008).Tracing the source of campylobacteriosis. PLoS Genet 26, e1000203.Sheppard, S. K., Dallas, J. F., Strachan, N. J., MacRae, M., McCarthy, N. D., Wilson, D. J., Gormley, F. J., Falush, D., Ogden, I. D., Maiden, M.C. & Forbes, K. J. (2009). Campylobacter genotyping to determine the source of human infection. Clin Infect Dis 48, 1072-1078.Mullner, P., Spencer, S. E., Wilson, D. J., Jones, G., Noble, A. D., Midwinter, A. C., Collins-Emerson, J. M., Carter, P., Hathaway, S. & French,N. P. (2009). Assigning the source of human campylobacteriosis in New Zealand: A comparative genetic and epidemiological approach. InfectGenet Evol. 138:1372-83.
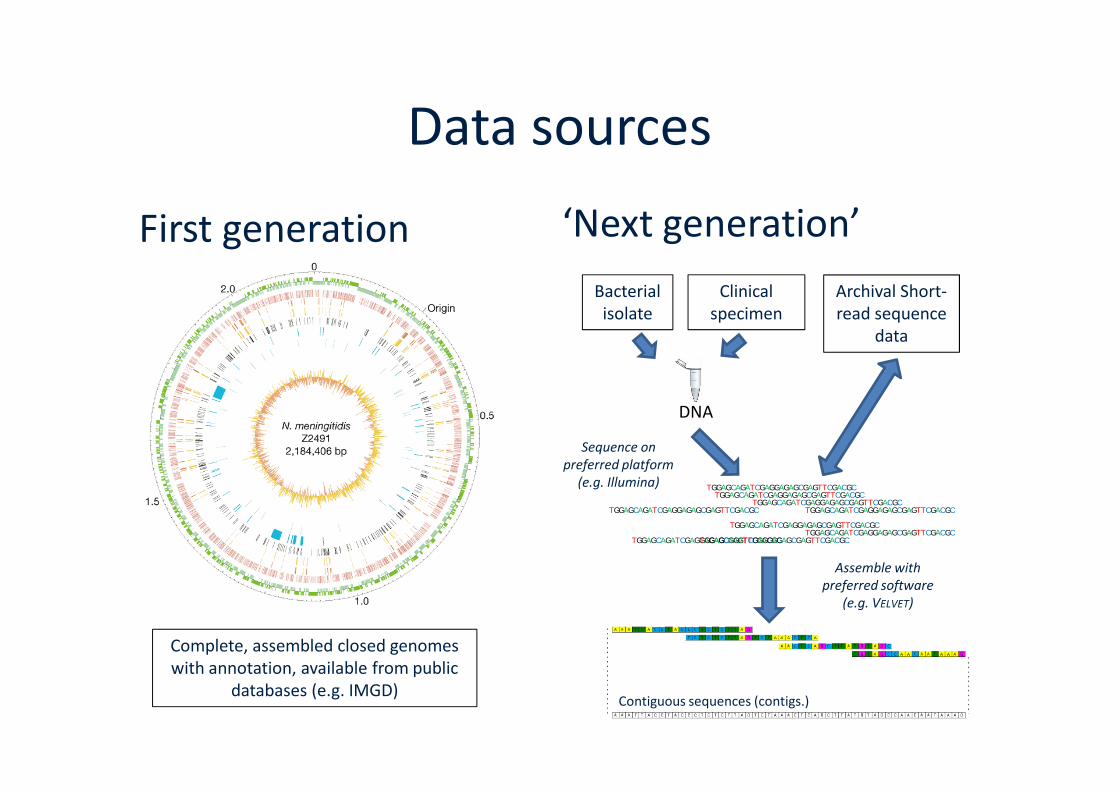
Data sources
First generation ‘Next generation’
Archival Short-read sequence
data
Bacterialisolate
Clinicalspecimen
Contiguous sequences (contigs.)
DNA
Sequence onpreferred platform
(e.g. Illumina)
Complete, assembled closed genomeswith annotation, available from public
databases (e.g. IMGD)
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGCTGGAGCAGATCGAGGAGAGCGAGTTCGACGC
Assemble withpreferred software
(e.g. VELVET)

Approaches to the analysis of wholegenome sequence data
• Comparison of short reads to a reference– ‘SNP’ calling: compares short reads to a high quality reference,
particularly used in comparing very closely related isolates.
• De novo assembly and comparison– ‘k-mer’ approach: reference-free assembly and comparison,– ‘k-mer’ approach: reference-free assembly and comparison,
independent of biological information.
• Gene-by-gene analysis– ‘whole genome MLST (wgMLST)’: de novo reference free or guided
assembly, followed by locus by locus identification and comparison ofgenetic variation.
Maiden, M. C., van Rensburg, M. J., Bray, J. E., Earle, S. G., Ford, S. A., Jolley,K. A. & McCarthy, N. D. (2013). MLST revisited: the gene-by-gene approach tobacterial genomics. Nat Rev Microbiol. 11(10): 728-36.
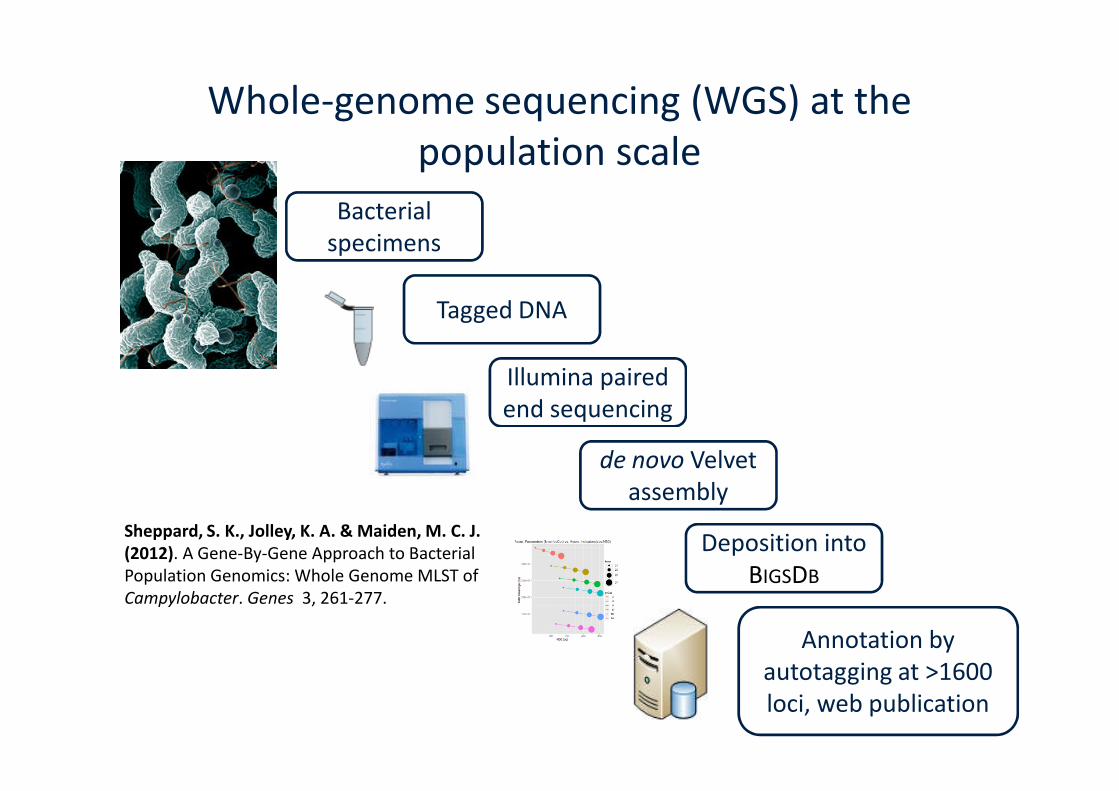
Whole-genome sequencing (WGS) at thepopulation scale
Bacterialspecimens
Tagged DNA
Illumina pairedend sequencingend sequencing
de novo Velvetassembly
Deposition intoBIGSDB
Annotation byautotagging at >1600loci, web publication
Sheppard, S. K., Jolley, K. A. & Maiden, M. C. J.(2012). A Gene-By-Gene Approach to BacterialPopulation Genomics: Whole Genome MLST ofCampylobacter. Genes 3, 261-277.
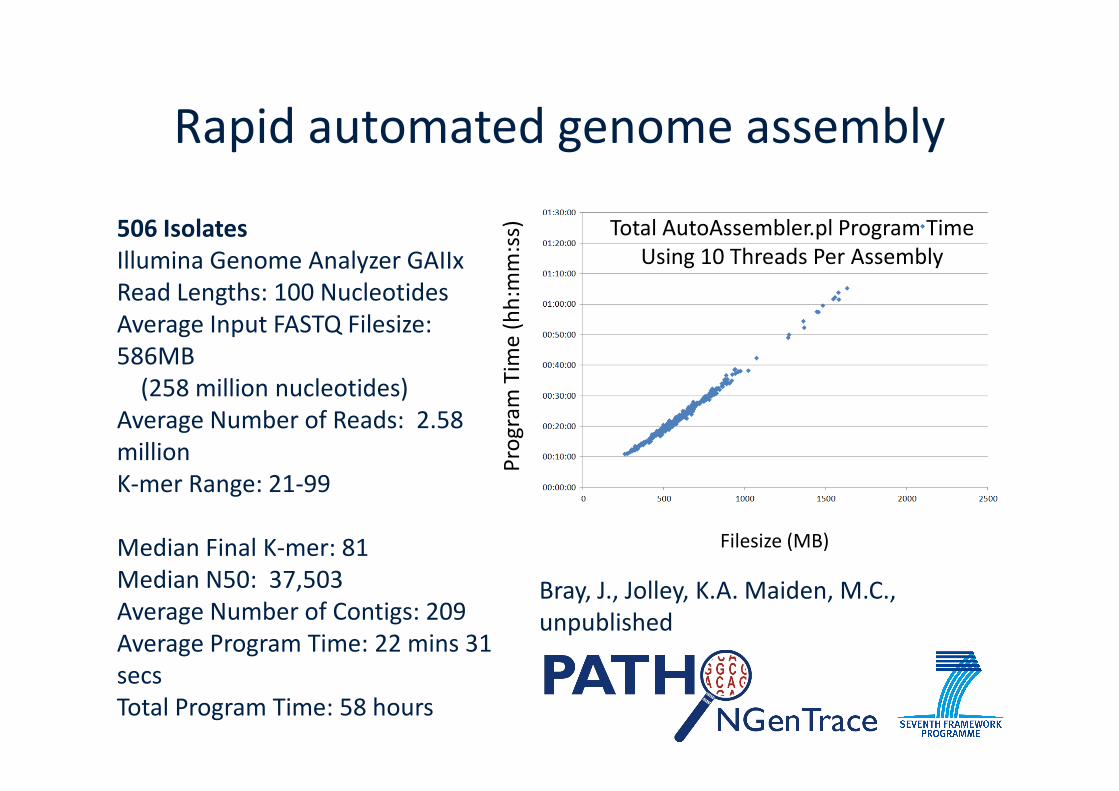
Rapid automated genome assembly
506 IsolatesIllumina Genome Analyzer GAIIxRead Lengths: 100 NucleotidesAverage Input FASTQ Filesize:586MB
(258 million nucleotides)
Pro
gram
Tim
e(h
h:m
m:s
s) Total AutoAssembler.pl Program TimeUsing 10 Threads Per Assembly
(258 million nucleotides)Average Number of Reads: 2.58millionK-mer Range: 21-99
Median Final K-mer: 81Median N50: 37,503Average Number of Contigs: 209Average Program Time: 22 mins 31secsTotal Program Time: 58 hours
Filesize (MB)
Pro
gram
Tim
e(
Bray, J., Jolley, K.A. Maiden, M.C.,unpublished
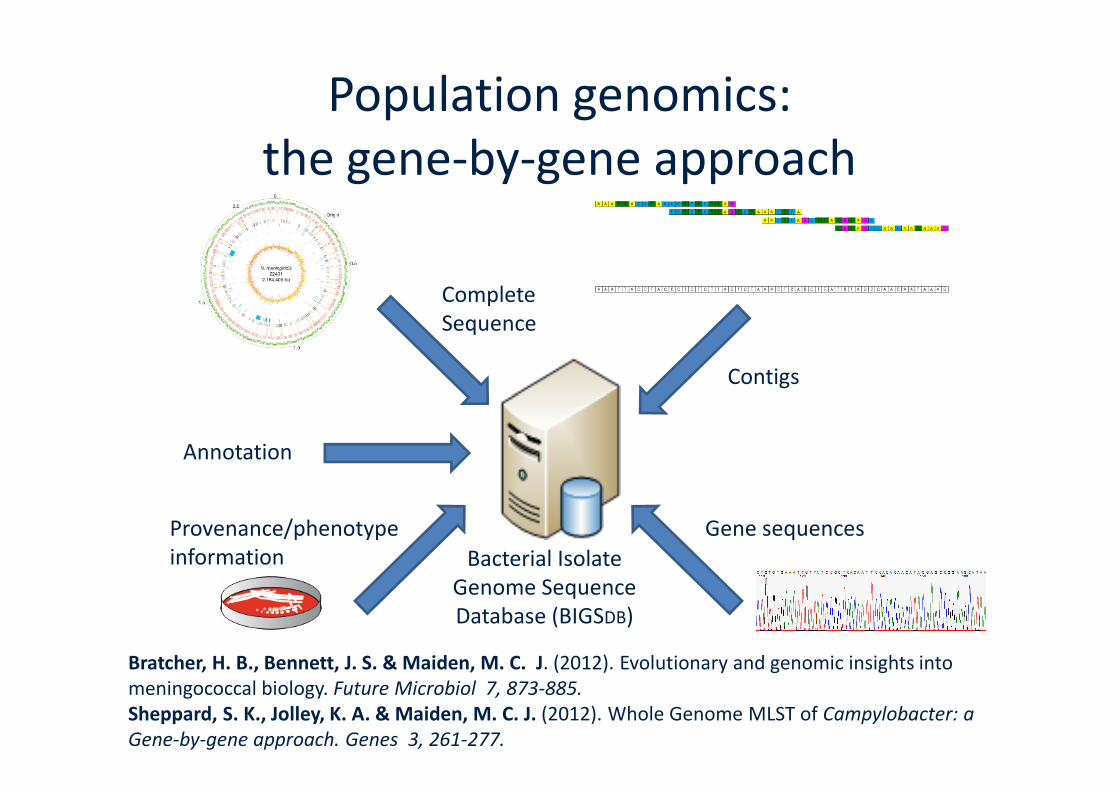
Population genomics:the gene-by-gene approach
CompleteSequence
Contigs
Annotation
Bacterial IsolateGenome SequenceDatabase (BIGSDB)
Gene sequencesProvenance/phenotypeinformation
Bratcher, H. B., Bennett, J. S. & Maiden, M. C. J. (2012). Evolutionary and genomic insights intomeningococcal biology. Future Microbiol 7, 873-885.Sheppard, S. K., Jolley, K. A. & Maiden, M. C. J. (2012). Whole Genome MLST of Campylobacter: aGene-by-gene approach. Genes 3, 261-277.

Bacterial Isolate Genome Sequence Database(BIGSDB)
CCATCCCGTTGTCGAACAGCAGGTACGCCA
CTTCACCGCCAACCACACCGACCTTGACCAC
AAACACCGCCTCATGCTGCTCACCGGCCCC
AATATGGGCGGCAAATCCACCTACATGCGCAGGAACCCTCAAAGCCGTTTTCCCGGAAAACCTATCCACAGCCGAACAGCTCCGCCAAGCCA
TTTTGCCCGAACCTTCCGTCTGGCTGAAAGACGGCAATGTCATCAACCACGGTTTTCATCCC
GAACTGGACGAATTGCGCCGCATTCAAAACCATGGCGACGAATTTTTGCTGGATTTGGAAGCCAAGGAACGCGAACGTACCGGTTTGTCCAC
ACTTAAAGTCGAGTTCAACCGCGTTCACGGCTTTTACATTGAATTGTCCAAAACCCAAGCCG
AACAAGCACCTGCCGACTACCAACGCCGGCAAACCCTTAAAAACGCCGAACGCTTCATCACGCCGGAACTGAAAGCCTTTGAAGACAAAGT
GCTGACTGCTCAAGAGCAAGCCCTCGCCTTAGAAAAACAACTCTTTGACGGCGTATTGAAA
AACCTTCAGACGGCATTGCCGCAGCTTCAAAAAGCCGCCAAAGCCGCCGCCGCGCTGGACGTGTTGTCCACATTTTCAGCCTTGGCAAAAG
AGCGGAACTTCGTCCGCCCCGAGTTTGCCG
ACAAGTCGCGCTGATTGTTT
AACCTTCAGACGGCATTGCCGCAGCTTCAAA
AAGCCGCCAAAGCCGCCGCCGCGCTGGACGTGTTGTCCACATTTTCAGCCTTGGCAAAAG
AGCGGAACTTCGTCCGCCCCGAGTTTGCCGACTATCCGGTTATCCACATCGAAAACGGCCGCCATCCCGTTGTCGAACAGCAGGTACGCCA
CTTCACCGCCAACCACACCGACCTTGACCACAGGAACCCTCAAAGCCGTTTTCCCGGAAAAC
CTATCCACAGCCGAACAGCTCCGCCAAGCCATTTTGCCCGAACCTTCCGTCTGGCTGAAAGACGGCAATGTCATCAACCACGGTTTTCATCC
CGAACTGGACGAATTGCGCCGCATTCAAAACCATGGCGACGAATTTTTGCTGGATTTGGAAG
CCAAGGAACGCGAACGTACCGGTTTGTCCACACTTAAAGTCGAGTTCAACCGCGTTCACGGCTTTTACATTGAATTGTCCAAAACCCAAGCC
GCCCCGAGTTTGCCGACTATCCGGTTATCCACATCGAAAACGGCCGCCATCCCGTTGTCGA
ACAGCAGGTACGCCACTTCACCGCCAACCACACCGACCTTGACCACAAACACCGCCTCATGCTGCTCACCGGCCCCAATATGGGCGGCAAA
TCCACCTACATGCGCCAAGTCGCGCTGATTGTTT
AGGAACCCTCAAAGCCGTTTTCCCGGAAAAC
CTATCCACAGCCGAACAGCTCCGCCAAGCCATTTTGCCCGAACCTTCCGTCTGGCTGAAAGACGGCAATGTCATCAACCACGGTTTTCATCC
CGAACTGGACGAATTGCGCCGCATTCAAAACCATGGCGACGAATTTTTGCTGGATTTGGAAG
CCAAGGAACGCGAACGTACCGGTTTGTCCACACTTAAAGTCGAGTTCAACCGCGTTCACGGCTTTTACATTGAATTGTCCAAAACCCAAGCC
GAACAAGCACCTGCCGACTACCAACGCCGGCAAACCCTTAAAAACGCCGAACGCTTCATCA
CGCCGGAACTGAAAGCCTTTGAAGACAAAGTGCTGACTGCTCAAGAGCAAGCCCTCGCCTTAGAAAAACAACTCTTTGACGGCGTATTGAAA
AACCTTCAGACGGCATTGCCGCAGCTTCAAAAAGCCGCCAAAGCCGCCGCCGCGCTGGAC
GTGTTGTCCACATTTTCAGCCTTGGCAAAAGAGCGGAACTTCGTCCGCCCCGAGTTTGCCGACTATCCGGTTATCCACATCGAAAACGGCCG
CCATCCCGTTGTCGAACAGCAGGTACGCCACTTCACCGCCAACCACACCGACCTTGACCAC
AAACACCGCCTCATGCTGCTCACCGGCCCCAATATGGGCGGCAAATCCACCTACATGCGCCAAGTCGCGCTGATTGTTT
abcZadkaroEfumCgdhpdhCpgmporAporB
Sequencebin
Locusdefinitions
tables: porBfetApenArpoB16SLocus XLocus Y
Jolley, K. A. & Maiden, M. C. (2010). BIGSdb: Scalableanalysis of bacterial genome variation at thepopulation level. BMC Bioinformatics 11, 595.
tables:annotation
source Locus Allele Provenance
abcZ 2 Country UK
adk 3 Year 2013
aroE 4 serogroup B
gdh 8 Disease carrier
pdhC 4 Age 23
pgm 6 Source Swab
... etc... ... etc ...

Genome annotation
• Genome-wide MLST
• Autotagger – runsregularly – tags all lociwith known alleles
• Each unique sequence• Each unique sequencegiven new allelenumber
• Loci grouped in toschemes
Sheppard, S. K., Jolley, K. A. & Maiden, M. C. J. (2012). A Gene-By-Gene Approach to Bacterial Population Genomics: Whole Genome MLSTof Campylobacter. Genes 3, 261-277.
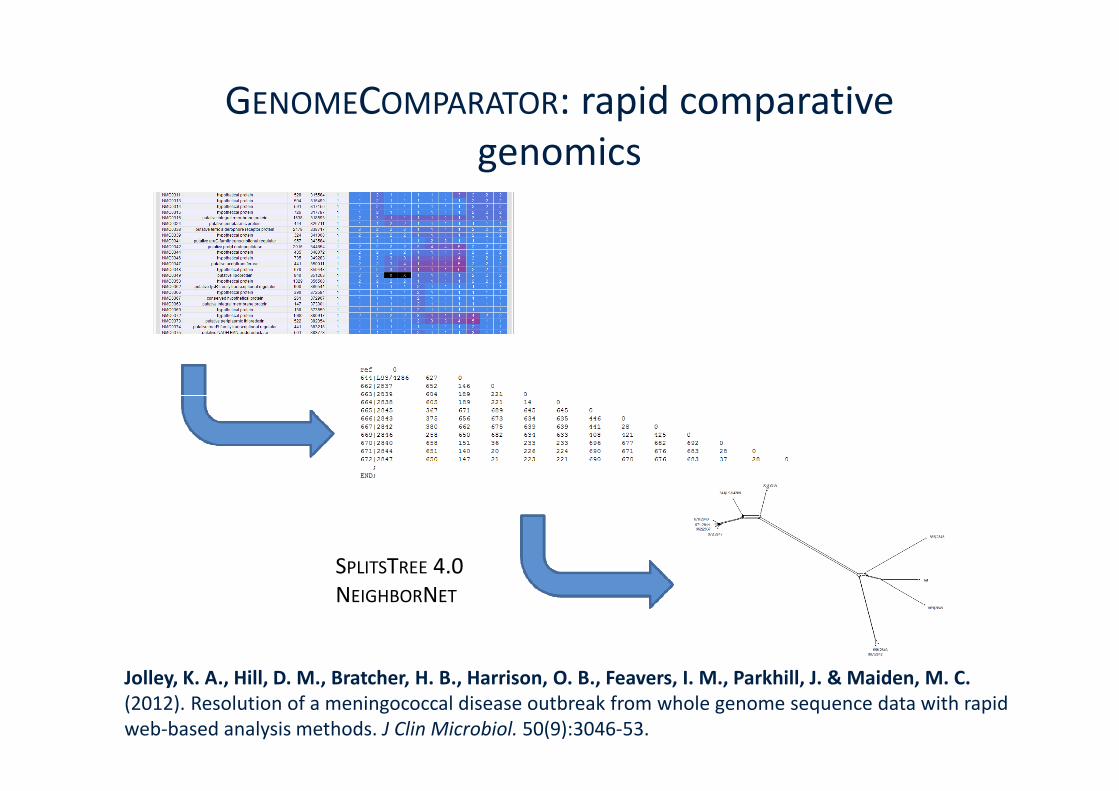
GENOMECOMPARATOR: rapid comparativegenomics
Jolley, K. A., Hill, D. M., Bratcher, H. B., Harrison, O. B., Feavers, I. M., Parkhill, J. & Maiden, M. C.(2012). Resolution of a meningococcal disease outbreak from whole genome sequence data with rapidweb-based analysis methods. J Clin Microbiol. 50(9):3046-53.
SPLITSTREE 4.0NEIGHBORNET

Ribosomal multi-locus sequencetyping, rMLST
• Isolate characterisation from ‘domain to strainfrom WGS data.
• Indexes the 53 universally presentribosomal genes.
• September 2015 ribosomal alleles defined for:• >129,791 genome sequences;
Jolley, K. A., Bliss, C. M., Bennett, J. S., Bratcher, H. B., Brehony, C. M., Colles, F. M.,Wimalarathna, H. M., Harrison, O. B., Sheppard, S. K., Cody, A. J. & Maiden, M. C. (2012).Ribosomal Multi-Locus Sequence Typing: universal characterisation of bacteria fromdomain to strain. Microbiology 158, 1005-1015.
• >129,791 genome sequences;• 1,169 genera;• 3,434 unique species ;
• rSTs defined for 25 taxa;• Neisseria and Campylobacter to clonal
complex and Salmonella to the Serovarlevel.
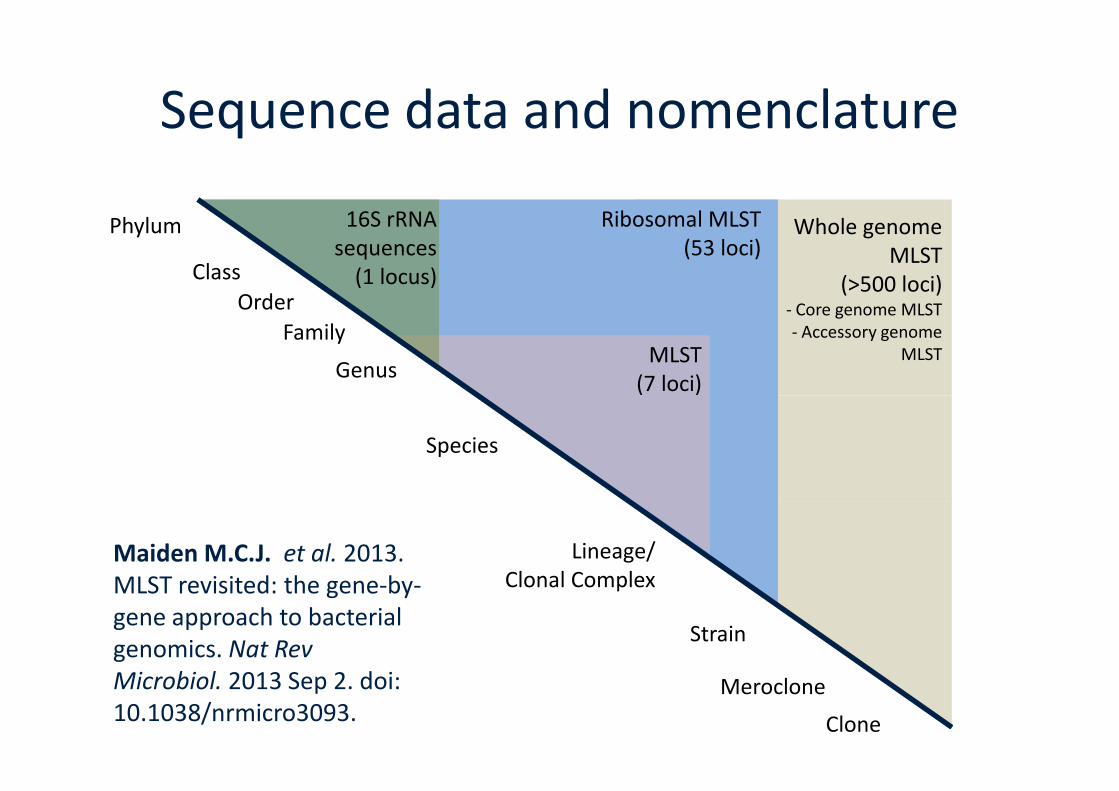
MLST(7 loci)
16S rRNAsequences
(1 locus)
Ribosomal MLST(53 loci)
Family
Order
Class
Phylum
Genus
Whole genomeMLST
(>500 loci)- Core genome MLST- Accessory genome
MLST
Sequence data and nomenclature
(7 loci)
Strain
Lineage/Clonal Complex
Species
Clone
Meroclone
Maiden M.C.J. et al. 2013.MLST revisited: the gene-by-gene approach to bacterialgenomics. Nat RevMicrobiol. 2013 Sep 2. doi:10.1038/nrmicro3093.

Data submitters:currently >1300;
Data curators:currently >90 MLST schemes
Sequence definitionsMLST, rMLST, antigengenes, core genome,pan-genome
Isolate datasets
• provenance• phenotype• gene content• allelic variation• genomes
Populationannotation
• locus classification• description• biochemical
pathway
Comparativegenomics
PubMLST1998*, 2003
Gene-by-gene analysisusing reference genomeor defined loci
Gen
eA
Gen
eB
Gen
eC
Gen
eD
Allele1: TTTGATACTGTTGCCGAAGGTTTAllele2: TTTGATACCGTTGCCGAAGGTTTAllele3: TTTGATTCCGTTGCCGAAGGTTT
>750 citations
• genomes
Linked to:
pathway• Core + accessory
genome analysis• Association studies
Molecular typingSpecies identification
EpidemiologyVaccine coverage/impact
Linking genotypeto phenotype
Outbreak investigationPopulation structure
>8000 unique visitors/month*http://mlst.zoo.ox.ac.uk
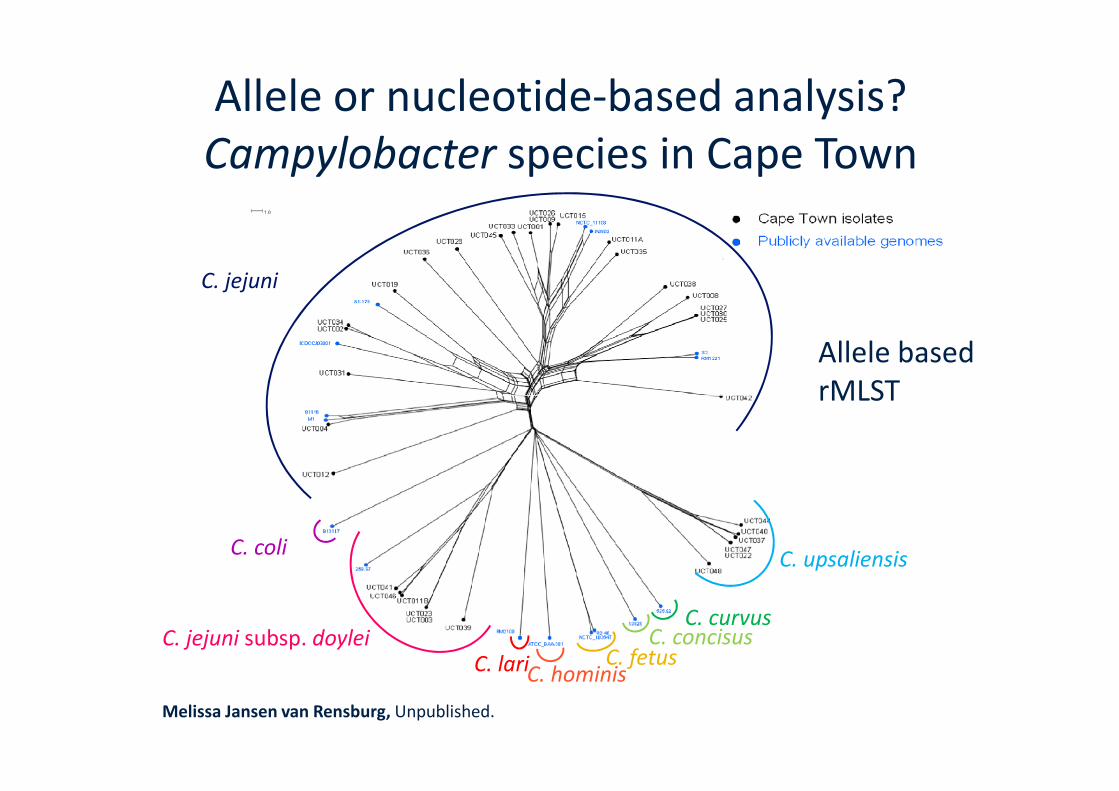
Allele or nucleotide-based analysis?Campylobacter species in Cape Town
C. jejuni
Allele basedrMLST
C. jejuni subsp. doylei
C. coli
C. lariC. hominisC. fetus
C. concisusC. curvus
C. upsaliensis
Melissa Jansen van Rensburg, Unpublished.
rMLST
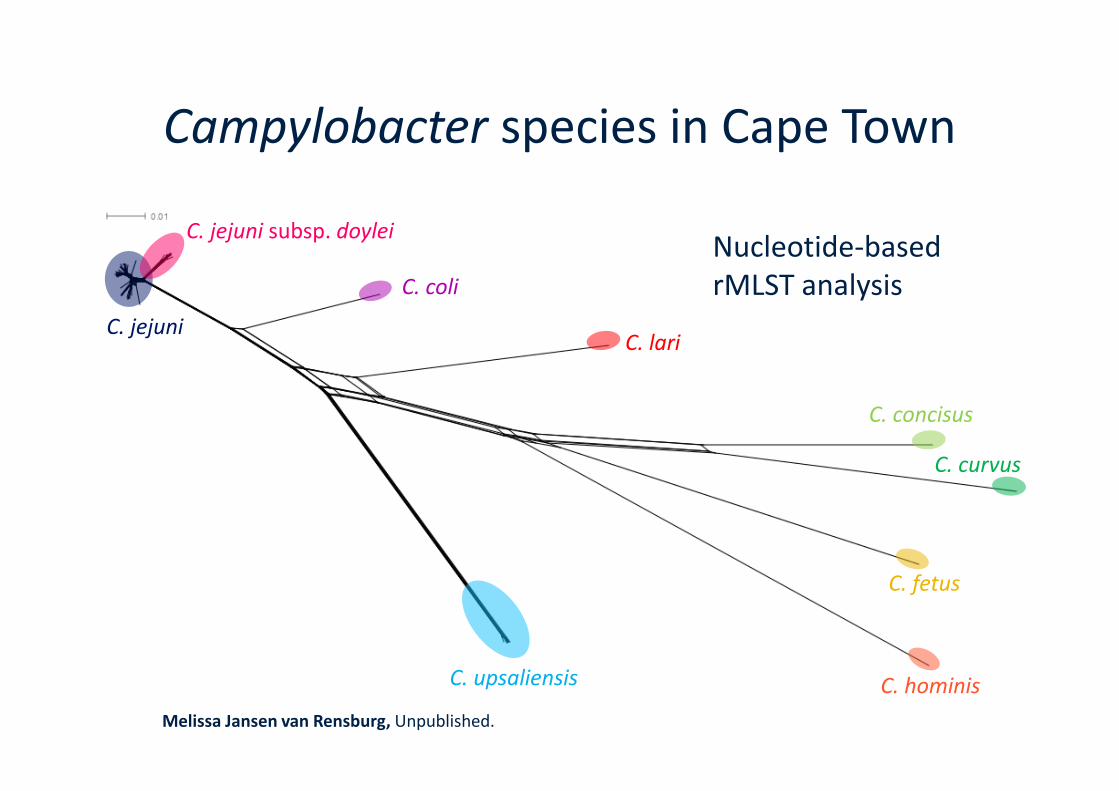
Campylobacter species in Cape Town
C. jejuni
C. jejuni subsp. doylei
C. coli
C. lari
Nucleotide-basedrMLST analysis
C. hominis
C. fetus
C. concisus
C. curvus
C. upsaliensis
Melissa Jansen van Rensburg, Unpublished.

Campylobacteriosis in Oxfordshire
• John Radcliffe Hospital Microbiologylaboratory: 800-900 Campylobacterisolates per year.
• Catchment area contiguous,Population ~600,000, about 1% of UKtotal.total.
• Ongoing surveillance since 2003:7,101 isolates (June 2015).
• Routine genomic surveillance ofCampylobacter isolates since June2011, 3,562 WGS (June 2015).
Cody, A. J., McCarthy, N. M., Wimalarathna, H. L., Colles, F. M., Clark, L., Bowler, I. C., Maiden, M. C. & Dingle, K. E. (2012). Alongitudinal six-year study of the molecular epidemiology of clinical Campylobacter isolates in Oxfordshire, UK. J Clin Microbiol 50,3193-3201.Cody, A. J., McCarthy, N. D., Jansen van Rensburg, M., Isinkaye, T., Bentley, S., Parkhill, J., Dingle, K. E., Bowler, I. C., Jolley, K. A. &Maiden, M. C. (2013). Real-time genomic epidemiology of human Campylobacter isolates using whole genome multilocus sequencetyping. J Clin Microbiol 51, 2526-2534.
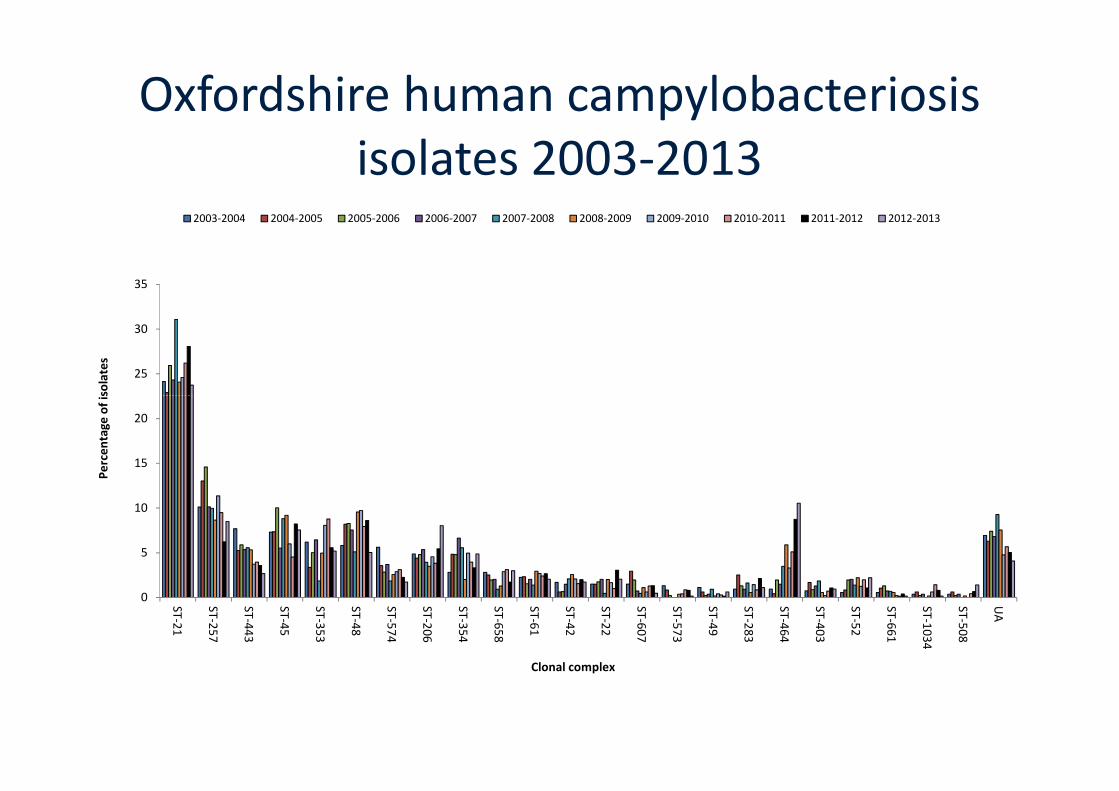
25
30
35
Pe
rce
nta
geo
fis
ola
tes
2003-2004 2004-2005 2005-2006 2006-2007 2007-2008 2008-2009 2009-2010 2010-2011 2011-2012 2012-2013
Oxfordshire human campylobacteriosisisolates 2003-2013
0
5
10
15
20
ST-21
ST-25
7
ST-44
3
ST-45
ST-35
3
ST-48
ST-57
4
ST-20
6
ST-35
4
ST-65
8
ST-61
ST-42
ST-22
ST-60
7
ST-57
3
ST-49
ST-28
3
ST-46
4
ST-40
3
ST-52
ST-66
1
ST-10
34
ST-50
8
UA
Pe
rce
nta
geo
fis
ola
tes
Clonal complex

STRUCTURE attribution of clinicalisolates from Oxfordshire 2003-2013
Retail chicken, yellow; cattle, dark blue; sheep, light blue; wild bird sources, brown.
Cody, A. J., McCarthy, N. D., Bray, J. E., Wimalarathna, H. M., Colles, F. M., van Rensburg, M. J., Dingle, K. E., & Maiden, M.C. Unpublished
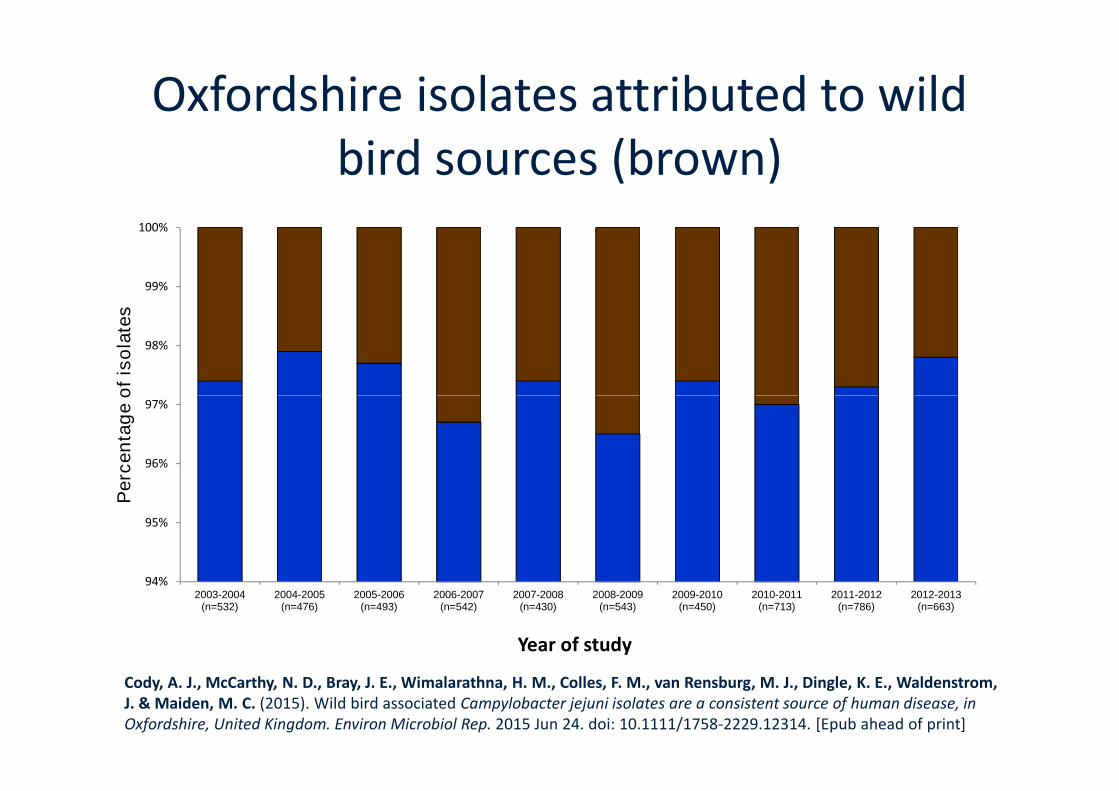
Oxfordshire isolates attributed to wildbird sources (brown)
98%
99%
100%
Perc
en
tag
eo
fis
ola
tes
94%
95%
96%
97%
2003-2004(n=532)
2004-2005(n=476)
2005-2006(n=493)
2006-2007(n=542)
2007-2008(n=430)
2008-2009(n=543)
2009-2010(n=450)
2010-2011(n=713)
2011-2012(n=786)
2012-2013(n=663)
Perc
en
tag
eo
fis
ola
tes
Year of study
Cody, A. J., McCarthy, N. D., Bray, J. E., Wimalarathna, H. M., Colles, F. M., van Rensburg, M. J., Dingle, K. E., Waldenstrom,J. & Maiden, M. C. (2015). Wild bird associated Campylobacter jejuni isolates are a consistent source of human disease, inOxfordshire, United Kingdom. Environ Microbiol Rep. 2015 Jun 24. doi: 10.1111/1758-2229.12314. [Epub ahead of print]
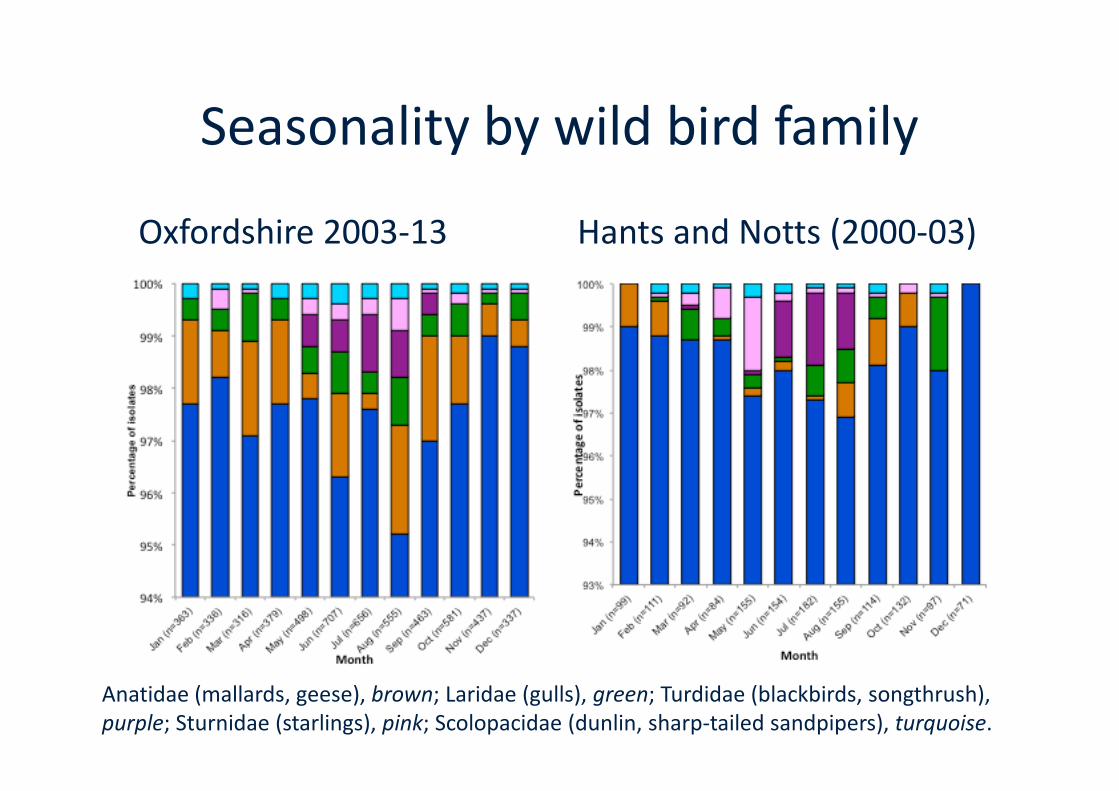
Seasonality by wild bird family
Oxfordshire 2003-13 Hants and Notts (2000-03)
Anatidae (mallards, geese), brown; Laridae (gulls), green; Turdidae (blackbirds, songthrush),purple; Sturnidae (starlings), pink; Scolopacidae (dunlin, sharp-tailed sandpipers), turquoise.
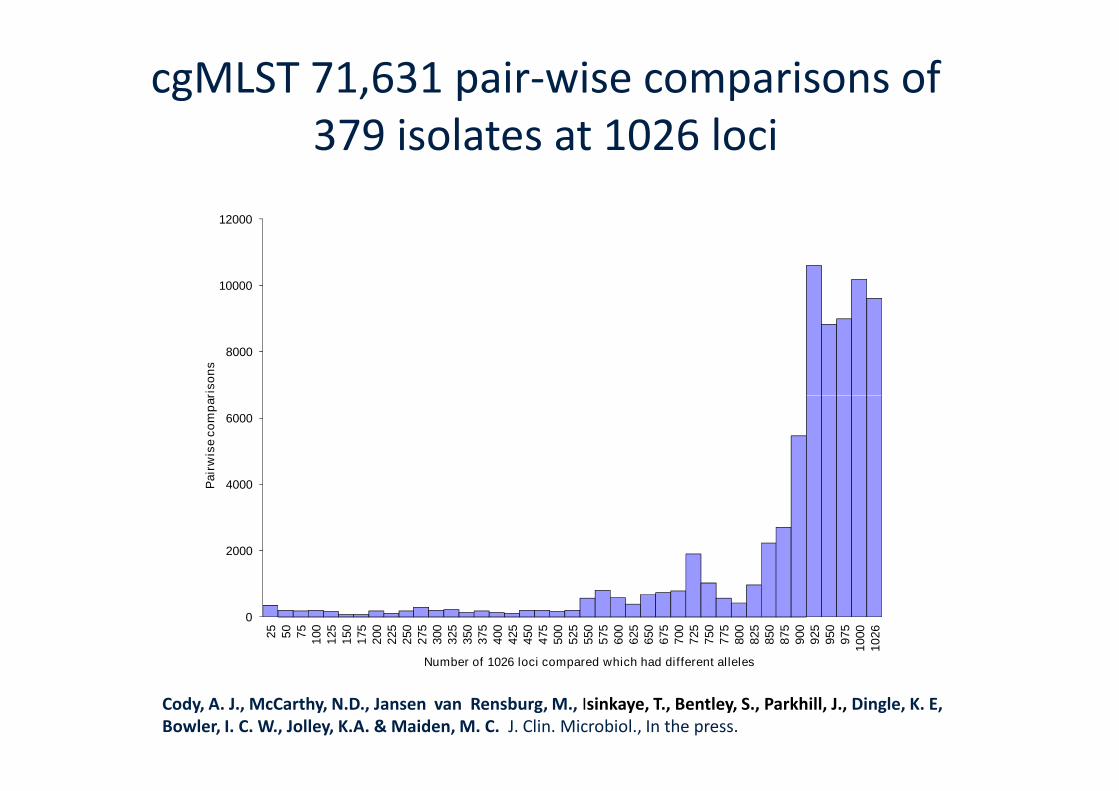
cgMLST 71,631 pair-wise comparisons of379 isolates at 1026 loci
8000
10000
12000P
air
wis
eco
mp
ari
so
ns
0
2000
4000
6000
25
50
75
100
125
150
175
200
225
250
275
300
325
350
375
400
425
450
475
500
525
550
575
600
625
650
675
700
725
750
775
800
825
850
875
900
925
950
975
1000
1026
Pair
wis
eco
mp
ari
so
ns
Number of 1026 loci compared which had different alleles
Cody, A. J., McCarthy, N.D., Jansen van Rensburg, M., Isinkaye, T., Bentley, S., Parkhill, J., Dingle, K. E,Bowler, I. C. W., Jolley, K.A. & Maiden, M. C. J. Clin. Microbiol., In the press.
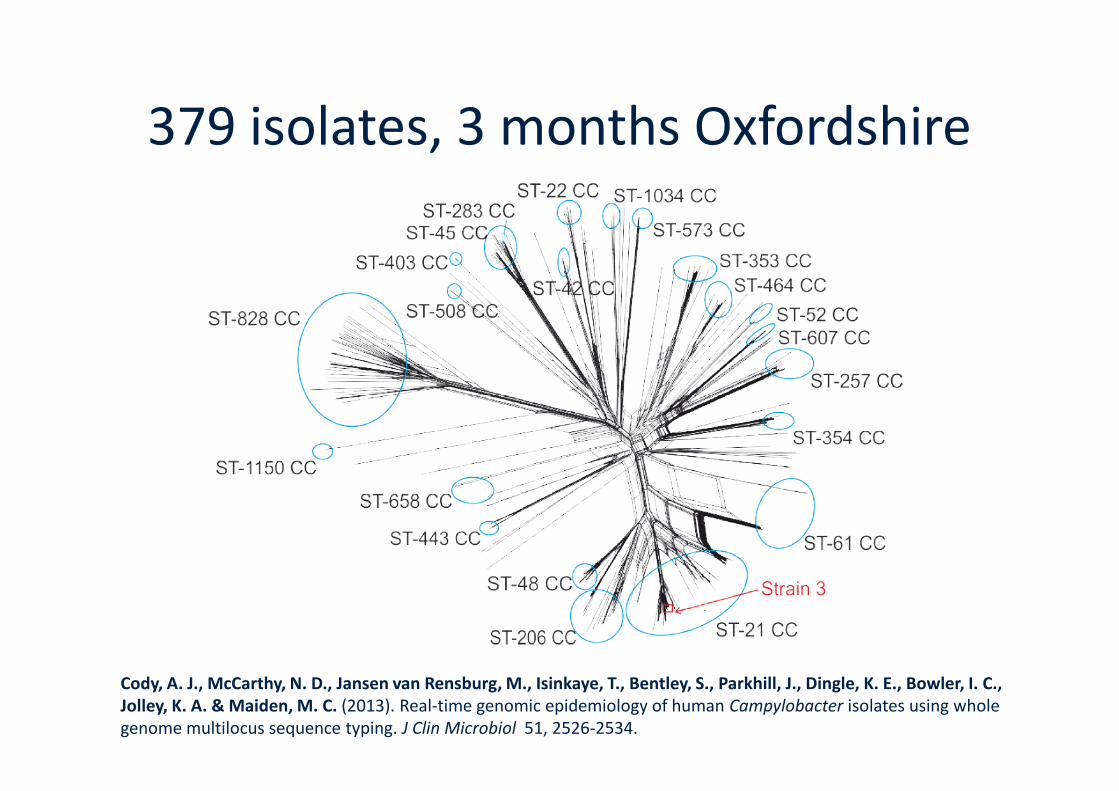
379 isolates, 3 months Oxfordshire
Cody, A. J., McCarthy, N. D., Jansen van Rensburg, M., Isinkaye, T., Bentley, S., Parkhill, J., Dingle, K. E., Bowler, I. C.,Jolley, K. A. & Maiden, M. C. (2013). Real-time genomic epidemiology of human Campylobacter isolates using wholegenome multilocus sequence typing. J Clin Microbiol 51, 2526-2534.

Global diversity of Campylobacter isolates,humans and animals over 10 years
ST-828 CC
ST-508 CC
ST-45 CC
ST-61 CCST-22 CC
ST-42 CCST-403 CC
ST-52 CC
ST-658 CC
ST-353 CC
ST-677 CCST-179 CC
ST-1275 CC
Lefebure, T., Pavinski Bitar, P. D., Suzuki, H. & Stanhope, M. J. (2010). EvolutionaryDynamics of Complete Campylobacter Pan-Genomes and the Bacterial Species Concept.Genome Biology and Evolution 2, 646-655.
ST-206 CC
ST-48 CC
ST-21 CC
ST-443 CCST-446 CC
ST-1150 CC

NEIGHBORNET allele-based wgMLST:‘Strain 3’ isolates
Cody, A. J., McCarthy, N.D., Jansen van Rensburg, M., Isinkaye, T., Bentley, S., Parkhill, J., Dingle, K. E,Bowler, I. C. W., Jolley, K.A. & Maiden, M. C. J. Clin. Microbiol., In the press.

NEIGHBORNET allele-based wgMLST:‘Strain 3A’ isolates
Cody, A. J., McCarthy, N.D., Jansen van Rensburg, M., Isinkaye, T., Bentley, S., Parkhill, J., Dingle, K. E,Bowler, I. C. W., Jolley, K.A. & Maiden, M. C. J. Clin. Microbiol., In the press.

Outbreak: single diverse source
Abid, M., Wimalarathna, H., Mills, J., Saldana,L., Pang, W., Richardson, J. F., Maiden, M. C. &McCarthy, N. D. (2013). Duck Liver-associatedOutbreak of Campylobacteriosis amongHumans, United Kingdom, 2011. Emerg InfectDis 19, 1310-1313.
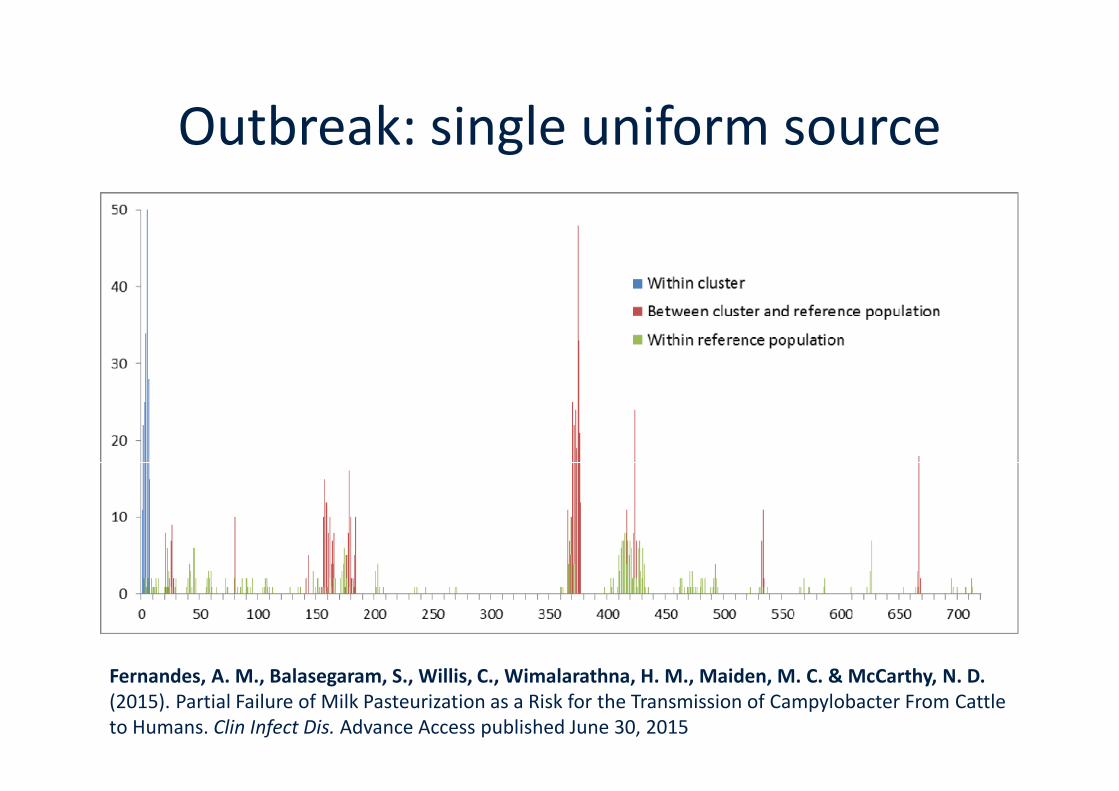
Outbreak: single uniform source
Fernandes, A. M., Balasegaram, S., Willis, C., Wimalarathna, H. M., Maiden, M. C. & McCarthy, N. D.(2015). Partial Failure of Milk Pasteurization as a Risk for the Transmission of Campylobacter From Cattleto Humans. Clin Infect Dis. Advance Access published June 30, 2015

Genomics in the courts...

Hierarchical gene-by-gene tracking ofCampylobacter
Centuries+ decades years months weeks days hours
Evolution emergence epidemiology diagnosis
OXC6289
OXC6393
OXC6459
Cluster 3
Relative discrimination required HighLow
Relative amount of genetic change LowHigh
OXC6565
OXC6527
OXC6530
OXC6543
OXC6524
OXC6636
OXC6590
OXC6461
OXC6393
OXC6598
OXC6600
Cluster 1
Cluster 2
Cluster 3