1.1 introduction - shodhgangashodhganga.inflibnet.ac.in › bitstream › 10603 › 4243 ›...
TRANSCRIPT

1 | Chapter 1
CRYSTAL ENGINEERING
1.1 Introduction Supramolecular chemistry1 is a new and rapidly progressing field on the
crossroads between chemistry, biology, physics and materials science. It is the chemistry
of molecular assemblies (beyond molecules) and of intermolecular interactions (non
covalent bonds). This not only provides the basis for revolutionizing numerous branches
of industry but also improves our understanding of the functioning of living organisms
and of the origin of life. Designing a new supramolecular system with desired properties
will provide us a better understanding about non-covalent interactions between
molecules within the molecular aggregates and it will transform the pharmaceutical
industry and medicine by developing new ways of drug administration and new
composite biocompatible materials which will serve as implants of new generation. The
existence of intermolecular forces was first postulated by Johannes Diderik van der
Waals in 1873. In the early twentieth century noncovalent bonds were understood in
greater detail, with the hydrogen bond being first described by Latimer and Rodebush2a
in 1920 and later by Linus Pauling in an extended treatment.2b,c The importance of
supramolecular chemistry was established by the 1987 Nobel Prize for Chemistry being
awarded to Donald J. Cram, Jean-Marie Lehn, and Charles J. Pedersen in recognition of
their work in the development and synthesis of shape and ion selective receptors or
"host-guest" complexes. Afterwards it took a rapid pace with the concepts of
mechanically-interlocked molecular architectures, crystal engineering and
supramolecular materials coming within its fold. In the 1990s, supramolecular chemistry
became even more sophisticated. The science of nanotechnology also had a strong
influence on the subject, with building blocks such as fullerenes, nanoparticles, and
dendrimers becoming part of synthetic systems.
CHAPTER
1

2 | Chapter 1
Within the realm of supramolecular chemistry another well-defined area is
crystal engineering,3 devoted to the design and studies of crystals built of two or more
components with desirable properties. Pepinsky first introduced the term “Crystal
Engineering” in 1955 and the subject was elaborated by Schmidt4 during 1950 to 1970 to
address the issue of crystal packing in the context of organic solid state photochemical
reactions of cinnamic acids and amides. A general meaning of the term was proposed by
Desiraju3a of Crystal Engineering as "the understanding of intermolecular interactions in
the context of crystal packing and the utilization of such understanding in the design of
new solids with desired physical and chemical properties". Crystal engineering is a
mainline interdisciplinary subject today that was started with organic solids and now
deals with the self-assembly of molecular crystals, metal–organic architectures,
nanostructures, and coordination polymers using hydrogen bonding, electrostatic, van
der Waals interactions, and metal coordination bonding. It is an interdisciplinary field
that seeks to develop protocols for predicting and controlling the structure and functional
properties of solids. Catalysis, optical materials, conducting and magnetic materials,
nanotechnology, electronic materials and sensors, nano and microporous materials,
supramolecular devices, protein-receptor binding, molecular modeling, drug design and
improving properties of existing APIs are some of the key research areas within the
realm of crystal engineering.
1.2 Intermolecular Interactions and Supramolecular Synthons
Crystal, the supramolecule par excellence,5 is an assembly of millions of
molecules held together in a periodic arrangement at an amazing level of precision by
intermolecular interactions, guided by molecular recognition and organized self
assembly. Intermolecular interactions include ion-ion, ion-dipole, dipole-dipole
interactions, hydrogen bonding, London forces, etc. The close packing principle of
Kitaigorodskii6 postulates that molecules in a crystal pack such that the projections of
one molecule dovetail into the hollows of its neighbour, i.e. bumps fit into hollows just
like lock and key, so that the maximum numbers of intermolecular contacts are achieved.
The crystal structure of a molecule is the free energy minimum resulting from the
optimization of several attractive and repulsive intermolecular interactions with varying

3 | Chapter 1
strengths, directional preferences and distance-dependence properties. Therefore
understanding the nature and strength of intermolecular interactions is of fundamental
importance in supramolecular chemistry. Intermolecular forces are mainly of two types,
(i) isotropic or non-directional (C···C, C···H, H···H interactions) that defines the shape,
size and close packing and (ii) anisotropic or directional3a as hydrogen bonds, charge
transfer interactions, halogen interaction, and heteroatom interactions (e.g. O–H···O, N–
H···O, C–H···O, C–H···N, O–H···π, halogen···halogen, nitrogen···halogen, sulfur···halogen
etc). Long range dispersion forces and short range repulsive forces are isotropic. These
interactions vary with r–n, where r is the distance between relevant non-bonded atoms
and n is a positive integer. The attractive forces vary from r–1 to r–6 depending upon the
interaction type and short range exchange repulsion varies with r–12. Among all
intermolecular interactions hydrogen bonding is the most reliable directional interaction
and it has a fundamental role in crystal engineering.3a Hydrogen bonds are classified into
three categories based on their strength as very strong, strong and weak hydrogen bonds
(Table 1).7 The properties of a crystalline material are the result of molecular
arrangement in the crystal lattice, which is controlled by intermolecular interactions.
Table 1 Some properties of very strong, strong and weak H-bonds.
Properties Very strong Strong Weak
Bond energy (–kcal mol–1)
15–40 4–15 <4
Examples [F···H···F]–
[N···H···N]+
P–OH···O=P
O–H···O=C O–H···O=C O–H···O–H
C–H···O O–H···π Os–H···O
Red shift in IR >25% 5–25% <5% D(X···A) (Å) 2.2–2.5 2.5–3.2 3.0–4.0 D(H···A) (Å) 1.2–1.5 1.5–2.2 2.0–3.0 θ(X–H···A) (º) 175–180 130–180 90–180 Covalency Pronounced Weak Vanishing Electrostatic Significant Dominant Moderate
From crystal engineering point of view the strong, directional forces are more
helpful to design target crystal structures. The interaction motifs for designing crystals

4 | Chapter 1
are termed as supramolecular synthons8a,b and Desiraju defined it as “supramolecular
synthons are structural units within supermolecules which can be formed and/or
assembled by known or conceivable synthetic operations involving intermolecular
interactions.” The concept is widely used in the design of solids which are important
from scientific and commercial viewpoints. The synthesis of supramolecular structures in
the solid state dealing with the self-assembly of molecular crystals using hydrogen
bonding, electrostatics, π-stacking, halogen bonding, van der Waals interactions, and
metal-coordination bonding. Crystal engineering is effectively like supramolecular
synthesis in the solid state, and there is a direct analogy between the supramolecular
synthon and the molecular synthon8c,d that was originally proposed for organic synthesis
by E. J. Corey8c in 1967. The advantage of using the synthon approach is that it offers a
simplification in the understanding of crystal structures. Zaworotko sub-classified
synthons as homosynthons and heterosynthons based on the interacting functional
groups. If supramolecular synthon is formed between the same functional group it is a
homosynthon, if it forms between two different functional groups it is called as
heterosynthon.9 Some of the well known homosynthons are COOH···COOH,
CONH2···CONH2, OH···OH, NH2···NH2, halogen···halogen, etc. which are between
similar functional groups and COOH···pyridine, CONH2···pyridine, COOH···CONH2,
OH···NH2, CONH2···N-oxide, halogen bonds, etc. are heterosynthons (Figure 1).
O
O H
O
O
H
O
N H
N
O
H N
O
H
O
O
H
H
O
O H N
H
Homosynthons Heterosynthons Figure 1 Examples of homosynthons and heterosynthons.
Similar to hydrogen bonds, halogen bonds10 are the noncovalent interaction
between halogen atoms (Lewis acids) and neutral or anionic Lewis bases, emerging as
prototype to hydrogen bonding. The interaction energy for halogen bond spans over a

5 | Chapter 1
wide range from 1 to 35 kcal mol–1. The weak Cl···Cl interaction between chlorocarbons
and the very strong I–···I2 interaction in I3– being the extremes. Weak interactions11 that
include C−H···O and C−H···N hydrogen bonds,11a C−H···π,11i halogen···halogen
interactions10 etc. are important in crystal design. C−H···O and C−H···N hydrogen bonds
are electrostatic in nature and have long-range distance character that have importance in
wide variety of chemical and biological systems. C–H···O hydrogen bonds are capable of
exhibiting all the properties11e that are similar to strong hydrogen bonds such as
dependence on the acidity and basicity of donor and acceptor strengths and near linearity
of the interaction and lone-pair directionality of the acceptor.
Identification of molecular functionalities that will generate predictable or robust
intermolecular interactions/ synthons is the key step of crystal engineering. The situation
will become more complicated in multi-functional molecules because of competition
between similar strength donor/acceptor groups. To understand hydrogen bonding and its
competition in organic compounds, Etter proposed12 three hydrogen bond rules as, (a)
“all acidic hydrogens available in a molecule will be used in hydrogen bonding in the
crystal structure of that compound,” (b) “all good proton acceptors will be used in
hydrogen bonding when there are available hydrogen-bond donors,” and the third rule is
that (c) “the best hydrogen-bond donor and the best hydrogen acceptor will
preferentially form hydrogen bonds to one another.” These rules provide useful
information about the preferred connectivity patterns, hydrogen bond competition and
stereoelectronic properties of hydrogen bonds for a particular functional group or for sets
of functional groups. The methods of ranking solid-state hydrogen bond preferences are
based on functional group competitions in homomeric crystals or heteromeric cocrystals.
These rules involved in analyzing which donors are selected by a limited number of
acceptors or vice versa during crystallization. The analysis of hydrogen bonds and other
weak interactions and its cooperation and competition will guide the construction of
target architectures and functions. A huge storehouse of crystal structures in the
Cambridge Structural Database (CSD)13 of nearly 4,83,000 crystal structures up to May
2009 (compared to very small sized 2000 entries in 1965) provides an excellent tool for
accessing the efficiency and reproducibility of a particular supramolecular synthon in
molecular crystals. A study on the probabilities of occurrence of supramolecular

6 | Chapter 1
synthons in a supramolecular system in presence or absence of second competing
functionality is discussed in Chapter 6 with a detailed chronological literature survey.
Our synthon analysis study14 of carboxylic acid, pyridine, amine, and hydroxyl
functional groups while present in same supramolecular system is discussed in Chapter
6.
Bernstein15 developed geometrical notations to recognize the hydrogen bond
patterns that are known as Graph Set Notation. Graph set approach is nothing but to
analyze the hydrogen-bond patterns from a complicated networks structure to a reduced
simple pattern. There are combinations of four, each specified by a designator: chains
(C), rings (R), intramolecular hydrogen-bonded patterns (S), and other finite patterns
(D). Specification of a pattern is augmented by a subscript designating the number of
hydrogen-bond donors d and a superscript giving the number of hydrogen-bond
acceptors a. In addition, the number of atoms n in the pattern is called the degree of the
pattern and is specified in parentheses. The graph set descriptor is then given as [G ad (n)],
where G represents one of the four possible designators. Some examples are given in
Figure 2.
OH
O
O H
12
3
45
6
O
N HH
O
NHH
1
2
34 5
6
78
O
N RH
1
2
3
4
O
N RH
C(4)
P OOH
R (8)2
2
S(6)
D
Figure 2 Examples of various graph set descriptors.
1.3 Organic Solid-State Forms
A crystal is "a three dimensional atomic, ionic, or molecular structure consisting
of periodically repeated, identically constituted, congruent unit cells"16a and the process
of the formation of solid crystals from the homogeneous solution, melt or by direct vapor

7 | Chapter 1
deposition is known as crystallization.16b Crystal is a well-defined pattern, or structure,
dictated by forces acting at the molecular level and during its formation process the
solute concentration should reach a certain critical value, before changing status
otherwise solid formation is impossible below the solubility threshold at the given
temperature and pressure conditions. Crystallization process consists of two main events,
(i) nucleation and (ii) crystal growth. Nucleation is the step where the solute molecules
dispersed in the solvent start to assemble into clusters, on the nanometer scale (elevating
solute concentration in a small region), that becomes stable under that conditions. The
stable clusters constitute the nuclei otherwise they re-dissolve and form the stable once
again. Supersaturation is the driving force for initial nucleation step. The nuclei are
stable only when they reach a critical size and such critical size is dictated by the
operating conditions (temperature, supersaturation, etc.). Single crystal X-ray diffraction
and powder X-ray diffraction are two very powerful techniques to determine crystal
structures. Crystal structures offer an understanding of various forces responsible for
holding the organic crystalline solids that can be engineered to have desired properties.
The nature of crystallization process is governed by thermodynamic and kinetic factors
(Figure 3). Several research groups studied crystal growth aspect. 17 Davey et. al.17a,b and
Desiraju et. al.17c studied nucleation and crystal growth on the crystal formation pathway
of tetrolic acid and Na(saccharinate).nH2O systems respectively. These are some of the
typical studies to understand primary stage of crystallization. Ostwald18 stated that a
system moves to equilibrium from an initial high-energy state through minimal changes
in free energy. Therefore the structure that crystallizes first is one which has the lowest
energy barrier (highest energy, kinetically metastable). This form would then transform
to the next lower energy polymorph until a thermodynamically stable state is reached,
the so-called Ostwald’s Law of Stages (Figure 4).

8 | Chapter 1
Figure 3 Hypotheitcal transitions from solution to thermodynamic and kinetic crystals. Small difference between ∆G ≠
thermodynamic and ∆G ≠
kinetic determines formation of kinetic
crystals.
Figure 4 Ostwald’s Rule of Stages. Initial high-energy state (metastable A) through minimal changes in free energy crystallizes first is one which has the lowest energy barrier. Metastable A form will then transform to the next lower energy polymorph (metastable B) and so on (metastable C) until thermodynamically stable crystal D.

9 | Chapter 1
Crystal engineering deals with various solid forms. It includes polymorphs, host-
guest complexes, network solids, salts, hydrates, cocrystals, more preferably
pharmaceutical cocrystals, and this chapter will cover a brief introduction to various
organic solid-state forms and the concept and its importance. When a compound
crystallized in two or more different crystalline modifications, they are known as
polymorphs and the phenomenon is polymorphism.19 Cocrystals20 can be defined as
multiple-component crystal structure in which two or more compounds coexist through
hydrogen bonds or non-covalent interactions. If the reactants are solids at ambient
conditions, the multi-component crystalline materials are cocrystals and those composed
of one or more solids and a liquid are known solvates or pseudopolymorphs21 and
hydrates22 when solvent is water. However the multi-component system is known as
molecular salt/ salt14,23 if proton is transferred from acid to base and retains as ionic state.
Thus salts and cocrystals are multicomponent crystals that can be distinguished by the
location of the proton between an acid and a base. Cartoon depictions of all these solid
phases are illustrated in Figure 5.
Figure 5 Cartoon depictions of various organic solid forms and discussed in the corresponding Chapters.

10 | Chapter 1
1.4 Polymorphism The word ‘Polymorphism’ originally comes from the Greek literature (poly =
many, morph = form). Polymorphism was first realized in 1798, when the German
chemist Martin Heinrich Klaproth discovered calcite minerals and aragonite had the
same chemical composition (CaCO3). Mitscherlich first documented polymorphism in
1822 in the context of crystallography24a of arsenate and phosphate salts that can exist as
different crystal forms. Ostwald’s work18 on the relative stability of different crystal
structures of the same compound was a major development in polymorphism.
Polymorphism acquired potential importance after Buerger and McCrone’s work24c on
change in properties like melting point and solubility of different crystal forms of the
same chemical compound. The existence of different crystal structures for the same
element or atom is known as allotrope first described by Berzelius.24d Allotropes are at
the elemental (e.g. C, S, P, Sn etc.) level whereas polymorphism is used to refer
structural diversity of molecular compounds.24e For example carbon has three allotropes,
diamond, graphite and fullerene. Depending on the atomic arrangement in their lattice
they show quite different properties. A widely accepted definition of polymorphism was
given by McCrone24f which states that “a solid crystalline phase of a given compound
resulting from the possibility of at least two crystalline arrangements of the molecules of
that compound in the solid state”. Buerger24g tried to simplify the definition limiting only
for solids composed of one component, but the concept was confusing and misleading as
polymorphism can exists in multi-component system as well.
Polymorphism may occur due to various reasons, packing of molecules,
conformational or molecular flexibilities, supramolecular synthon competitions are the
main reasons and subsequently they are called as packing, conformational and synthon
polymorphs19,25 (Scheme 1). Packing polymorphism exists when the molecule is mostly
rigid. Conformationally flexible molecules have greater scope for their polymorphic
occurrence because of large number of degrees of freedom as the energy differences
between conformational polymorphs lies in a small window of 0.5-3 kcal mol–1. A
metastable conformation may be stabilized by stronger hydrogen bonds in the crystal
structure while a stable conformer may not be able to form strong hydrogen bonds,
although they lead to a balance of energy state and the overall stability of a polymorph is
accounted by measuring conformation energy and lattice energy. The energy

11 | Chapter 1
compensation towards overall energy of a polymorphic system and the phenomenon
known as systematic effect was recently reviewed by Nangia25c with several examples of
conformational polymorphs. Our result on conformational and synthon polymorphism is
discussed in Chapter 2 and 4.
Conformational Polymorphs
Packing Polymorphs
Conformational Polymorphs
Synthon Polymorphs
Packing polymorphs
Conformational Isomorphs,
Polymorph i Polymorph ii
Polymorph iii
Polymorph iv
v
Polymorph vi Polymorph vii
cisoid
transoid
Scheme 1 Schematic illustrations of different arrangement of molecules in the crystalline lattice that leads to different kinds of polymorphism. This scheme is culled from A. Nangia Acc. Chem. Res. 2008, 41, 595.

12 | Chapter 1
Different polymorphs have different physical and chemical properties (Table 2).
Thus characterization of all polymorphs through polymorph screening using various
methods like solvent less methods of melt and sublimation via green methodology,
solution crystallization etc. and then identification of stable form and control over the
preparation of that particular form has become a major goal for academic (crystal
engineering and solid-state chemistry) and industry research. 19,26a Polymorphism of
drugs is of central interest after the Norvir and Zantac26 incidents in the last decade.
Dissolution profile of Ritonavir polymorphs shows a significant difference with unique
crystal structures. Polymorph I is almost five times more soluble compared with
Polymorph II which is almost insoluble. Thus a thorough screening and complete
characterization of all possible polymorphs is considered an essential step in
pharmaceutical industry to choose the best drug formulation with desirable properties.
The extensive study on polymorphism gives fundamental understanding on molecular
recognition, crystal nucleation, and structure–property relationships.27 Among the
various methods of polymorph generation, solution crystallization and/or high-
throughput crystallization28 are default. Recent approaches for polymorph generation
include crystallization with structurally related additives,29a,b epitaxial growth,29c laser
induced nucleation,29d crystallization in capillaries,29e confinement within porous
materials,29f using polymers as heteronuclei,29g,h mechanical grinding,29i using
supercritical liquids,29j using self assembled monolayer with different functional
moieties,29k potentiometric cycling29m etc. Recently melting and sublimation, the two
solvent less high temperature techniques to afford guest free host structures were
explored25d by our group and those techniques were employed to generate new
polymorphs of compound that are prone to give guest included crystal on solution
crystallization with very good probabilities.

13 | Chapter 1
Table 2 Properties that can be different for polymorphs. This table is culled from, S. Dutta, D. J. W. Grant, Nat. Rev. Drug Discovery, 2004, 3, 42.
Packing properties
• Molar volume and density
• Refractive index, optical properties
• Conductivity,electrical and thermal
• Hygroscopicity
Kinetic properties
• Dissolution rate
• Rates of solid state reactions
• Stability
Surface properties
• Surface free energy
• Interfacial tensions
• Habit
Mechanical properties
• Hardness
• Tensile strength
• Compactibility, tabletability
• Handling, flow and blending
Thermodynamic properties
• Melting and sublimation temperatures
• Internal energy
• Enthalpy
• Heat capacity
• Entropy
• Free energy and chemical potential
• Thermodynamic activity
• Vapour pressure
• Solubility
Spectroscopic properties
• Electronic transitions, ultraviolet-visible
spectra
• Vibrational transitions, infrared and
Raman spectra
• Rotational transitions
• Nuclear magnetic resonance chemical
shifts
1.5 Enantiotropic and Monotropic Related Polymorphs
The thermodynamics of polymorphs of molecular crystals can be represented by
pressure-temperature or commonly used energy-temperature phase diagram that are
helpful for characterizing and understanding polymorphic behavior of a compound.
Polymorphism exists in the solid state and natural physical process phase transition
between polymorphs is a common phenomenon. The stability relationship of polymorphs
of a molecule can be established by measuring their enantiotropic or monotropic
relationship.9a The two polymorphic modifications are said to be enantiotropic when the
transition point between the two phases is found at a temperature below the melting point
of either of them (Figure 6a). When there is no transition point below the melting point

14 | Chapter 1
of the two polymorphs then the two forms are monotropically related (Figure 6b). This is
known as heat-of-transition rule.9a The heat-of-fusion rule states that in an enantiotropic
system higher melting polymorph will have the lower heat of fusion. If the higher
melting polymorph has a higher heat of fusion the two polymorphs are monotropically
related. Solid and liquid will be in equilibrium at melting point and Gibbs free energy
will be zero for two phases. The entropy of fusion can be expressed as,
∆Sf = ∆Hf / Tf
Entropy of fusion rule states that two modifications are enantiotropically related
if polymorph with higher melting point has the lower entropy of fusion and
monotropically related if lower melting polymorph has lower entropy of fusion. eg. for a
dimorphic system (Figure 6a) it is seen that the thermodynamic transition point Tp,I/II
defined by the point at which GI and GII cross, falls at a temperature below the melting
point of lower melting form, mpII and hence enantiotropically related. However the free
energy curves do not cross at a temperature below the two melting points (Figure 6b) and
so they are monotropically related.
(a) (b) Figure 6 (a) Fundamental E/T diagram for dimorphic enantiotropic system. Form I is stable below transition point. Above transition point Form II is stable. (b) Fundamental E/T diagram for dimorphic monotropic system. Form I is more stable at all temperature below melting point than form II.
Several analytical techniques are being used to establish the thermodynamic
behaviour of polymorphs, eg. Optical and/ or Hot Stage Microscopy (HSM), Differential
Scanning Calorimetry (DSC) etc. HSM can be used to obtain qualitative information on
polymorphic behaviour however thermal analysis (DSC or DTA) provides quantitative

15 | Chapter 1
information about the relative stability of polymorphic modifications, the energies
involved in phase changes between them and the monotropic and enantiotropic nature of
those transitions. Tolbutamide [1-butyl-3-(4-methylphenylsulfonyl)urea], an oral
hypoglycaemic agent exists in four polymorphic modifications. Polymorph I is stable
and phase transition from polymorph III, II and IV can clearly be described by DSC
thermograms (Figure 7). Transitions III→I, II→I, IV→I show those pairs are
enantiotropically related. Monotropic and enantiotropic relation between polymorphs in
several instances are observed and discussed in Chapter 2 and 4 with the help of HSM,
DSC and X-ray diffractions. For characterization of polymorphs spectroscopic methods
that include infrared (FT-IR), near infrared (NIR) and Raman spectroscopy etc., thermal
analysis (DSC, TGA, HSM etc) and finally X-ray diffraction (single crystal and powder
X-ray diffraction) are used.
Figure 7 DSC thermogram of Tolbutamide polymorphs: (a) Form I, (b) Form II, (c) Form III and (d) Form IV. The polymorph conversions from other forms to Form I are shown. These transitions are enantiotropically related. 1.6 Isostructurality
Geometrical properties like shape, size and chemical like electronegativity,
polarizability of functional group influence crystal packing. Kitaigorodskii has given

16 | Chapter 1
importance to the volume and shape of functional groups in crystal packing,6 however
the electronic properties of functional groups cannot be over looked.30a,b As the size of
the molecule increases the significance of geometric effects becomes important. A given
packing motif may be able to tolerate small changes in the molecular structure without a
considerable change in the close-packed crystal structure. These changes are minor
alterations in substitution and/or epimerization. The tolerance may be ascribed to the
presence of ∼30% free space in close-packed structures because the packing coefficients
of organic crystals are generally about 70%.30d The phenomenon by which different
molecules pack in a similar fashion to produce similar crystal structures is called
isostructurality and the structures are called isostructural30e and is inversely related to the
phenomenon of polymorphism. Isostructurality and Isomorphism are two commonly
used terms in literature. Two crystals are said to be isomorphous31a if (a) they have the
same space group and unit-cell dimensions and (b) the types and the positions of atoms
in both are same except for a replacement of one or more atoms in one structure with
different types of atoms in the other (isomorphous replacement), such as heavy atoms, or
the presence of one or more additional atoms in one of them (isomorphous addition). The
substances are so closely similar that they can form a continuous series of solid solutions.
On the other hand, two crystals are said to be isostructural31b if they have the same
structure, but not necessarily the same cell dimensions nor the same chemical
composition, and with a comparable variability in the atomic coordinates to that of the
cell dimensions and chemical composition. e.g. calcite (CaCO3), sodium nitrate (NaNO3)
and iron borate (FeBO3) are isostructural. The phenomenon of isomorphism is known for
more than two centuries with the growth of potassium alum crystals from a saturated
solution of ammonium alum. Kitaigorodskii6 was the first to review isostructurality in
organic molecular crystals. Kálmán et al. have divided isostructurality into two
categories⎯isostructural crystals or main-part isostructuralism of related molecules and
homeostructural crystals and proposed two descriptors to quantify isostructurality. They
are unit-cell similarity index Π and isostructurality index Ii(n)30e and can be defined as
the following equations. When the related molecules differing by substitutions on more
than one atomic site have similar packing, it is called homeostructural crystals.

17 | Chapter 1
01 ≅−′+′+′
++=Π
cbacba
where a, b, c and a', b', c' are orthogonalized lattice parameters of the related structures.
For a pair of completely isostructural crystals Π should be close to zero.
1001)(
2/12
×⎥⎥⎥
⎦
⎤
⎢⎢⎢
⎣
⎡
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛ ∆−=∑
n
RnI i
i
i
The isostructurality index, [Ii(n)] is a measure of the degree of internal isostructurality
where n is the number of distance differences (∆Ri) between the absolute coordinates of
identical non-hydrogen atoms within the same section of asymmetric units of related
structures. Ii(n) should be close to 100% for isomorphous crystals.
Isostructurality in three dimesions means the complete crystal packing. However
one- and two dimensional isostructurality32 is documented. When two structures show
similar infinite two-dimensional molecular arrangements they are called as two-
dimensionally isostructural. Accordingly similar arrangement of molecules in 1D is one-
dimensionally isostructural. It is important to have some knowledge about which groups
are interchangeable and under which circumstances to see the isostructural behavior
between two structures. Kitaigorodskii33 has ranked them as, (i) the halogens Cl, Br, I;
(ii) O and S; (iii) C, quadrivalent Si, Ge, Sn and Pb. There are some examples where
strong hydrogen bonding functional groups, such as –OH, –NH2, =O, can also replace
hydrogen to produce isostructural crystals (Figure 8). Isostructurality phenomenon was
investigated for steroids by Kálmán with an exchange of functional groups
(gamabufotalin/arenobufagin) or by epimerization (5α- and 5β-androstane-3 α,17β-
diol).34 2-oxa-4-androstene-3,17-dione is isostructural with 6α-hydroxy analogue,
replaced C−H···O interaction by C−O−H···O hydrogen bond is an example of 1D
isostructurality from our group (Figure 8b).32b,c The fact that these two compounds form
solid solution validates the isomorphous replacement of C–H atom by C–OH group
(Figure 8d, 8e).

18 | Chapter 1
N
N
RMe
O2N O
O
O
R
O
O
R
OHH
OH
OH
H
H
R=H, NH2
(a)
R=H, OH
(b)
R=O, H2
(c)
H HH HH H
O O O
6
3
HOH
HOH
HO
H
O O O3
6
(d) (e)
Figure 8 (a) H/NH2 exchange forms isostructural crystals. (b) In 6α-Hydroxy-2-oxa-4-androstene-3,17-dione, H/OH exchange produces isostructural crystals. (c) H2/O exchange generates isostructurality. (c) via C−O−H···O synthon in (d) without disturbing the overall arrangement of molecules; (d) Hydrogen bonding of 2-oxa-4-androstene-3-17-dione when R = H and (e) R = OH. Identical a-axis and the similarity in hydrogen bonding and arrangement of molecule in both structures lead to isostructural, due to replacement of C−H···O synthon by C−O−H···O.
Triiodoresorcinol and triiodophloroglucinol are rare case of examples which are
both polymorphic and isostructural recently reported from our group.35a They crystallized
as orthorhombic (P212121) and monoclinic (P21/n) polymorphs and the orthorhombic
polymorphs of both compounds are isostructural and correspondingly monoclinic
polymorphs are also identical. These examples illustrate isostructurality via C–H ⇔ C–
OH replacements. Another example that shows both polymorphism and isostructurality
is 2-amino-4-chloro-6-morpholinepyrimidine and 2-amino-4-chloro-6-piperidino
pyrimidine.35b Exceptions are also reported. For example room temperature form of 2,6-
dichloro-N-phenylformaide and 2-chloro-6-methyl N-phenylformaide (both
orthorhombic) are isomorphous35c but their high temperature forms (both in monoclinic)
are not isomorphous. 2-Amino-4-chloro-6-morpholinopyrimidine is dimorphic with Z′ =
2 and 1 in space group P21/c. The Z′ = 2 polymorph is isostructural with 2-Amino-4-
chloro-6-piperidinopyrimidine that has only one crystal structure with Z′ = 2 showing O

19 | Chapter 1
⇔ CH2 replacement. Chloro–methyl exchange is well known and this rule states that
when the geometry of the groups governs the crystal packing they produce isostructural
crystals due to their similar size and shape (Cl 20 Å3 and Me 24 Å3),35d discussed
thoroughly and calculated isostructurality index and unit cell similarity index of newly
synthesized series of similar phenylbenzenesulfonamides and its polymorph structures
that is covered in Chapter 4. Isostructurality in organic solids is well documented in
literature.35 There is a report on bromide and nitrate exchange in isostructural crystals in
spite of their different shapes where both of the anions make strong H-bonds with the
cation counter part.35e Above all these groups, the halogen exchange,35f specially Cl, Br
and I to produce isostructurality, are more frequent. Propargylammonium halides (Cl¯,
Br¯, I¯) are isostructural where halide ions accept three H-bonds from ammonium group
and one from terminal alkyne group. Another interesting isostructurality has been
reported by Bar et al.35g in para sustituted X–C6H4–CH=N–C6H4–X' molecules. When X
= X' = Cl or Br, the molecules are not isostructural, but molecule with X = Cl and X' =
Br is isostructural to the dichloro compound. On the other hand X = Br and X' = Cl
substituted molecule is isostructural to dibromo derivative. It indicates the importance of
halogens as well as the position of substitution in the molecules. In principle, two
isostructural compounds are expected to yield the similar polymorphs35q and the idea is
similarly applicable for multi-component systems like solvate, salt and cocrystals etc.
Isostructural crystals often lead to similar kind of properties.35r,s
1.7 Polymorphism and High Z' Structures in Solvent Less Methods
Awkward molecular shape, OH, NH2, SO3H like sticky functional groups, ionic
nature etc. are some of the factors for hydration and/or solvent inclusion complexes of
organic molecules, especially APIs. In drug industry, solvent inclusion complexes are
not advisable because of the toxic vapor nature of most solvents. Thus the synthesis and
characterization of guest-free crystalline forms has gained importance but difficult to
crystallize because, in general a solvent or water molecule acts as a crystallization aid or
filler in the voids of the host. Methods that have been used to obtain guest free structure
of lattice inclusion host compounds and discussed in Chapter 2. Temperature lowering,
isothermal evaporation and isothermal diffusion crystallization techniques are common

20 | Chapter 1
methods to grow single crystals. Some popular methods to produce guest free host
crystal structure are (a) misfit size and/or shape36a of the guest molecule to the void
formed by the host, (b) using an appropriate dual-nature solvent or unfavourable
electrostatic interactions,36b,c (c) layer-by-layer conversion of particles from the outside
in as guest is leached out,36d (d) recrystallization with a solvent-nonsolvent36e system, (e)
sonication,36f (f) gradual pH change36g etc. High temperature crystallization methods
melting and sublimation36h,i were explored recently from our group. The two high
temperature solvent less methods of green methodology generate guest free structures of
well known host 1,1-bis-(4-hydroxyphenyl)cyclohexanej and further illustrated with
isomeric dihydroxybenzoic acid molecules and successfully isolated guest free forms and
new polymorphic modification of other cases are covered.
Z' (Z prime)37a,b is the number of symmetry independent or crystallographic
unique molecules in a crystal lattice. Structure with Z' = 1 means that each molecule is
surrounded by like molecules, however, Z' > 1 structure means each molecule is
surrounded by molecules that are crystallographically different. The occurrence and
reasons behind high Z' structures have attracted attention of crystallographers and now
being intensely studied to understand the factors leading to high Z′ crystal structures evev
as occurrence of high Z' structures is still not properly understood. Steed37a showed
presence of pseudosymmetry, awkward shape, formation of molecular helices via
hydrogen bond or other interactions, strong hydrogen bonds, chirality, kinetic or
temperature effect are different reasons for high Z' structures are elaborately discussed in
Chapter 3. Our observation25d is the frequent occurrence of high Z' structures in high
temperature solvent less methods of melt and sublimation and compared with Cambridge
Structural Database (CSD). It is found that solvent-free crystallization methods show a
much higher probability of multiple Z' structures (~18%) compared to overall CSD
trends on Z' frequencies (<12%). Generation of high Z' structures by melting and
sublimation crystallization can be understood as rapid cooling of the hot liquid or vapor
in the open flask or on the cold finger is a kinetic phase and the conditions under which
hydrogen-bonded clusters are likely to condense in a pseudo-symmetric crystalline
arrangement. Popular host 1,1-bis-(4-hydroxyphenyl)cyclohexane is found to be a
remarkable example to illustrate the occurrence of high Z' structure in metastable
polymorph by melting. Solvent less methods when used to generate guest free host

21 | Chapter 1
structures of isomeric dihydroxybenzoic acids, Z' > 1 structure is observed commonly
discussed in Chapter 2 and 3. Carbamazepine37c is another exciting example for which Z'
= 1 from solution crystallization (3 polymorphs) whereas Z' = 4 when it is crystallized
from melting.
1.8 Salt Cocrystal Continuum It is a must to label and classify crystalline solid forms in order to characterize
them and then make comparisons. There has been a long standing and lively debate on
the nomenclature issues in crystal engineering, starting from what is a cocrystal, or co-
crystal/salts, to the definition of pseudopolymorph, solvate, host–guest compounds etc.
In general, molecular crystals can be classified broadly into single-component and
multiple-component crystals. Salts and cocrystals are multi-component crystals there
exists a continuum linking cocrystals and salts based on the extent of proton transfer
between the components. Cocrystals can be defined as multiple-component crystal
structure in which two or more compounds coexist through hydrogen bonds or non-
covalent interactions. If the reactants are solids at ambient conditions, the multi-
component crystalline materials are cocrystals and those composed of one or more solids
and a liquid are known solvates or pseudopolymorphs. 21 However the multi-component
system is known as molecular salt/ salt if proton is transferred from acid to base in the
ionic state. If a solution containing an organic acid and an organic base deposits a
crystalline solid containing both components, the result can be a molecular salt or a
cocrystal. If the proton resides on the base, then proton transfer has occurred and the
crystalline acid-base complex is a molecular salt. If proton transfer has not occurred and
the proton remains on the acid, then it is a cocrystal.21,38a The propensity of an acid to
give up a proton is represented by its pKa, the negative logarithm of the dissociation
constant. pKa relates to the equilibrium behavior in aqueous solution and measured pKa
values will vary depending on measurement technique, solvent, temperature, and other
factors. The extent of proton transfer depends on the magnitude of the difference of pKa
values of the reacting acid and base. It is generally accepted that reaction of an acid with
a base will be expected to form a salt if the ∆pKa [∆pKa = pKa(base) – pKa(acid)] is
greater than 3.75, which is an essential criteria while selecting the appropriate counter
ions to the preparation of salts of API in order to improve its properties like solubility.

22 | Chapter 1
For acid-base complexes with similar pKa values the ∆pKa value and the crystalline
environment determine the extent of proton transfer. Johnson and Rumon studied38b the
type of hydrogen bonding interaction as a function of ∆pKa, where ∆pKa refers to the
difference in pKa, of pyridinium ion (BH+) and the benzoic acid (AH) in water via
infrared spectra of solid state complexes of benzoic acid and substituted benzoic acids
with pyridine and substituted pyridines. Extensive study by Nangia et. al.39 based on the
analysis of several cocrystals and salts, concluded that the carboxylic acid–pyridine O–
H···N interaction will be neutral when ∆pKa < 0 and it will have an intermediate H bond
character, O–H···N and/or N+–H···O–, when the transition range 0 < ∆pKa < 3.75. The
interaction will be ionic N+–H···O– when ∆pKa > 3.75 (Scheme 2). Similar observation
was noted by Childs and Stahly38 in their analysis of 20 complexes of theophylline with
COOH partners, which resulted in 16 salts, 2 cocrystals and 2 mixed ionizations states
with transition range 0 < ∆pKa < 2.5.
O
O
H N
O
O
H
O
O
NH
NO
O
HN
+−
+
Cocrystal
Salt
Mixed ionization state
I
II
III
∆pKa = pKa (pyrNH+) – pKa (COOH)
∆pKa < 0, neutral synthon (I), O–H···N
0 < ∆pKa < 3.75, mixed ionization
state, O···H···N (II) or I/III, O–H···N/
N+–H···O–
∆pKa > 3.75, ionic N+–H···O–
Scheme 2 The pKa rule thumb to predict the H-bonding motifs in multi-component crystals.
Although the contribution from Aakeroy, Black, Price, Tocher etc.39 is worthy, it
is really difficult to predict any general conclusions about proton transfer in acid-base
systems. A detailed discussion is recently reported from our group and presented in
Chapter 6. The pKHB scale, proposed by Laurence,40 measures the free energy of
hydrogen bonded complex (1.364 pKHB = –∆GHB in kcal mol–1) could be a better guide in
predicting H-bond pairing compared to pKa values as it deals with sharing of H atom

23 | Chapter 1
between two electronegative atoms, while the pKa scale considers only the ability of the
proton to be transferred from acid to base. The pKHB values are quite sensitive to factors
that modify H-bonding ability, e.g. inductive/resonance effects, steric hindrance, lone-
pair repulsion, and intramolecular H-bonding.
1.9 Pharmaceutical Cocrystals
Cocrystals and salts are very useful in designing extended supramolecular
architectures; prepare NLO materials, solid-state photodimerisation reactions, enantio
separation of racemic compounds, pharmaceuticals developments etc. Cocrystallization
is a very important technique to develop new pharmaceutical phases of active
pharmaceutical ingredients (APIs). Pharmaceutical cocrystals are crystalline molecular
complexes of an Active Pharmaceutical Ingredient (API) with another pharmaceutically
acceptable molecule or Generally Regarded As Safe (GRAS) chemicals. Food additives,
preservatives, excipients, vitamins, minerals, amino acids, bio-molecules, and other APIs
can be selected as cocrystal formers (CCF). Zaworotko et al. stated41 that polymorphs,
pseudopolymorphs, salts, molecular complexes and cocrystals of APIs can modify
chemical and physical properties that may lead to extended patent coverage and
consequent legal protection of products. Several pharmaceutical crystals are known to
undergo a variety of phase transformations. Phase transformations during processing and
formulation can affect the stability and bioavailablity of drugs. Crystalline APIs are
strongly preferred due to their relative ease of isolation, the rejection of impurities
inherent to the crystallization process and the physico-chemical stability that the
crystalline solid state affords. Crystal engineering affords a paradigm for rapid
development of APIs, that of pharmaceutical cocrystals and salts which can be rationally
designed. Recent articles42 emphasize the development and importance of
pharmaceutical cocrystals. For example, cocrystallization of aspirin, rac-ibuprofen, and
rac-flurbiprofen with 4,4′-bipyridine by Zaworotko;42a Fluoxetine hydrochloride with
pharmaceutically acceptable carboxylic acids (Figure 9b) by Childs;42b several drug
molecules with Saccharine as API saccharinate salts by Desiraju;42c,d Itraconazole with
1,4-dicarboxylic acids by Remenar,42e Carbamazepine with Saccharin as saccharinate
salts42f etc. were the well known strategies to deal with inadequate solubility, dissolution
24 | Chapter 1
rate, absorption, physical stability, complexation etc. of APIs. Extremely water insoluble
nature of Itraconazole, an antifungal agent, is solved by making itraconazole–succinic
acid cocrystals (Figure 9a) as oral formulation. Cocrystal of carbamazepine and
saccharin (CBZ–SAC) appears to be superior to existing crystal forms of CBZ with
respect to stability, favourable dissolution, suspension stability, and favourable oral
absorption profile. Hydration behaviour of caffeine and theophylline was controlled by
their 1:1 cocrystals with oxalic and other diacids. These cocrystals or salts exhibit
physical properties different from those of the parent compounds as a direct result of
hydrogen-bonding interactions between the binary components of the crystals. However,
the utility of cocrystal formers in pharmaceutical products is limited by their
pharmacological and toxicological properties.
Cocrystallization of polymorphic APIs may provide a route to obtain a single
pharmaceutical phase by controlled formation of specific supramolecular synthons
between functional groups. For example, Cocrystals of a polymorphic drug Piracetam
and Gentisic acid, p-hydroxybenzoic acids as cocrystal formers which are also
polymorphic and APIs were synthesized via acid-amide heterosynthon (Figure 9c).42i
The cocrystals do not exhibit polymorphism. However polymorpism in cocrystals or
multi-component systems is not so uncommon.43 A recent study from our group25b
showed43 there are 33 cocrystal polymorph sets up to the January 2008 release of the
CSD when compared to more than 1600 polymorphic systems of single component
crystals with our own results on cocrystal polymorphs of Temozolamide and bipyridine-
N-oxide. The cocrystal former strategy is being applied for the optimization of the drug
design, processing, and delivery procedures.
(a) (b)

25 | Chapter 1
(c) (d) Figure 9 (a) Structure of cis-itraconazole and succinic acid cocrystal. Succinic acid molecule is closely fitting between the two itraconazole molecules via O−H···N hydrogen bonding. (b) Hydrogen bonds between fluoxetine cations, benzoic acids and chloride ions in the cocrystals of fluoxetine hydrochloride with benzoic acid to improve physical properties of fluoxetine. (b) Cocrystals of piracetam with gentisic acid via acid-amide heterosynthon to control polymorphism. (d) Caffeine-glutaric acid cocrystals to solve hydration.
Salts and cocrystals have the potential to be much more useful in pharmaceutical
products than solvates or hydrates. But it is also true that making cocrystal or salt may
have adverse effects on physiological systems. For example nearly 4000 deaths of pets
occurred due to the renal failure is due to the additive in food. Melamine–cyanuric acid
cocrystal was given as protein additive.44 Investigations concluded the presence of
cyanuric acid as another co-contaminant along with melamine causes intratubular
precipitation of cocrystal leading to the kidney failure and the death of animals. Crystal
engineering of melamine and cyanuric acid (1:1 molar ratio) cocrystals show two-
dimensional networks in the solid-state that is highly insoluble in water and causes
immediate precipitation, which was the reason for deaths of animals.
1.10 Hydrates and Host Guest Compounds Hydration of molecules in the crystal structure is a common phenomenon,
especially in pharmaceutical industries. Hydrated structures received considerable
attention because of its different topologies in the structure, conformations and functions.
Hydrates are commonly used in pharmaceutical solids because of its abundance,
flexibility, small size and ability to act as both a strong hydrogen bond donor and
acceptor and obviously its non toxic nature.45 The study of different water clusters is also
important to understand the bulk properties of water and its role in different biological
processes, such as protein–DNA binding, ion transport, protein folding–defolding,

26 | Chapter 1
structure determination of the fibrous proteins, etc. Included water molecules can form
discrete and extended motifs, e.g. finite and infinite chains, ring motifs and different
topologies. In biological systems water channels have been found and water topology is
widely studied because of its application in water and ion transport.46 In red blood cells
and the renal tubules water can rapidly and selectively cross the plasma membrane. Thus
water release from the interface, in general, is favored entropically but enthalpically
unfavorable. Infantes and co-workers47 showed that 6.6% of organic compounds are
hydrated and this value increases to 75% for bioactive pharmaceutical compounds or
APIs and categorized and given rank for different functional groups that promote
hydration. Molecules containing charged or strong H-bonding functional groups favor
entrapment of waters into its crystalline lattice. Many fundamental biological processes
depend on potentially important water chains.48 Water chain motif is responsible in
proton transport in Gramicidin-A.48d Buchanan,48e Ripmeester48f and others have studied
water chains that can serve as a model for biological proton wires or water transport.
Henry showed that water can act also as templating nanoporous material.49 Due to the
difficulties in studying the role of water molecules in macromolecular systems,
entrapment of water in small molecular environment and then study has become an
interesting topic in recent solid state supramolecular chemistry.
Davy’s discovery50 of chlorine hydrate in 1811 can be recognized as the origin
of host-guest chemistry as well supramolecular chemistry. But the field took a rapid pace
after the seminal contribution from Busch, Curtis, Jägar, Pederson, and then Lehn’s work
towards host–guest compounds in the development and synthesizing shape and ion
selective receptors with macrocyclic ligands (Figure 10). The host-guest relationships
involve a complementary stereoelectronic arrangement of binding sites between host and
guest. The host component is defined as an organic molecule or ion whose binding sites
converge in the complex and the guest component are any molecule, ion whose binding
sites diverge in the complex. Host guest chemistry has received particular interest
because of their diverse applications in chemical separation, reactions and catalysis in a
microcavity, and for electrooptic, nonlinear and magnetic materials.51 Hydrogen bonds or
other weak interactions mediated self-assembly and directional metal−ligand
coordination bonding are used to synthesize porous materials or low density
frameworks.52

27 | Chapter 1
Busch (1964) Curtis (1961) Jäger (1964) Pedersen (1967)
Figure 10 The macrocyclic ligands are synthesized by Busch, Curtis, Jager and Pedersen.
Host-guest compounds are mainly divided into Cavitands and Clathrands based
on the nature of the host. Cavitands are intra-molecular cavities however clathrands are
hosts with extra-molecular cavities resulted from aggregation of more than one
molecule.51c Based on the size of porosity, open frameworks are divided into three
categories, such as nanoporous or microporous (<15 Å), mesoporous (15−500 Å) and
macroporous (>500 Å) materials.51a,h The cavities formed by host molecules can either
be of zero dimensional (cage), one dimensional (channel) or two dimensional (layered).
To design porous solids various methods have been developed based on crystal
engineering principles and hydrogen bonds or metal coordination bonds. Weber rules53
for designing host framework are discussed in Chapter 5. Bulky shape, rigid framework
structure, strong and directional bonding properties are some requirements to construct
host-guest crystals.
1.11 Network Solids
The rational construction of novel open-framework organic solids8 has received
considerable attention because of their diverse applications. One of the main challenges
in the approaches of constructing host-guest compounds is to prevent interpenetration to
obtain open frameworks. Selection of suitable building blocks is must to construct a
particular architecture. In supramolecular chemistry building architectures is important
and the molecular building blocks are known as “molecular tectonics” defined by
Wuest.54 The word “tecton” is taken from Greek for “builder.”
28 | Chapter 1
O H
O OH
OO H O
HOR
COOHHOOC
COOHHOOC
COOH
OH
OH
OH
OH
COOH
COOH
COOH
HOOC
COOHHOOC
O OH
OH
Diamondoid Net
Hexagonal (6,3) Net
Ladder Net
Hexagonal Net (6,3) Net
Zig Zag or Crinkled Tape
Chain or Linear Tape
Ladder Netand Hexagonal (6,3) Net
2D Herringebone (6,3) Net
Linear
V-Shaped
Y-Shaped
T-Shaped
H-Shaped
Tetrahedral
Molecule Tecton Network
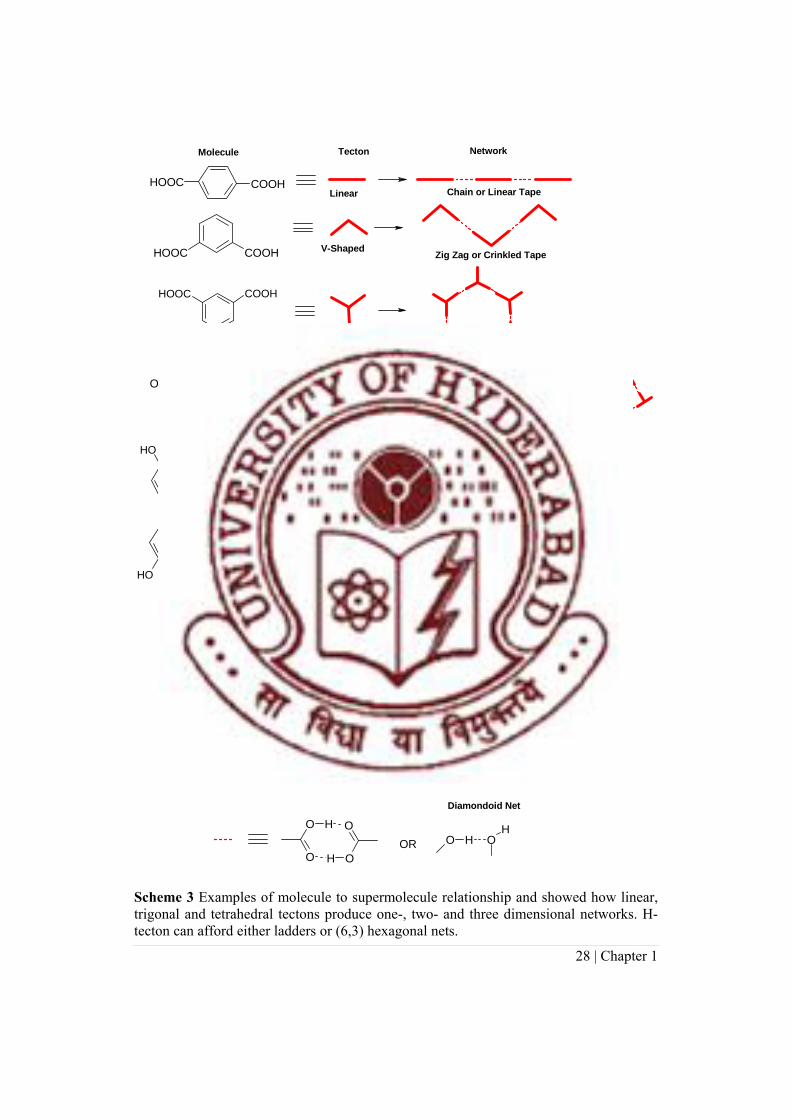
Scheme 3 Examples of molecule to supermolecule relationship and showed how linear, trigonal and tetrahedral tectons produce one-, two- and three dimensional networks. H-tecton can afford either ladders or (6,3) hexagonal nets.

29 | Chapter 1
This approach is a modular, programmed build up from molecule to crystal—rod
type molecules form linear aggregates, chiral and C2-symmetry molecules lead to helical
networks, C3/D3 symmetry molecules produce honeycomb grid or hexagonal layer
structures, and Td/S4 symmetry tectons self-assemble as adamantane or diamondoid
networks.55 H-shaped 1,4-di[bis(4′-hydroxyphenyl)methyl]benzene and its CH3 and
CH3O derivatives are synthesized and used to construct a diverge network topologies
recently reported from our group.56 The ability to predict the network architecture from
the shape and symmetry of the functionalized tecton is fundamental to crystal design. For
instance benzoic, terephthalic, trimesic and adamantane-1,3,5,7-tetracarboxylic acids
produce zero-, one-, two- and three-dimensional supramolecular structures respectively,
based on molecular geometry and carboxylic acid dimer synthon (Scheme 3). Our results
of ladder networks, (6,3) hexagonal network, rare pentagonal tiling by superposition of
two (6,3) hexagonal net, interpenetration and catenation are discussed in Chapter 5.
1.12 Conclusions
Single-component crystals (polymorphs) and multi-component crystals (salts,
solvates, hydrates, cocrystals and their polymorphs) are equally important to modify the
physical and chemical properties of drugs. Unsolvated forms are advisable because most
solvents are toxic and volatile in nature. Melt and sublimation are two high temperature
solvent less methods explored by our group to find guest free structures of those
compounds that are prone to give solvates upon solution crystallization. Thus a thorough
screening of all possible forms of API is considered to be very important step in
pharmaceutical industry. Single crystal X-ray diffraction, powder XRD diffraction, FT-
IR, NIR, Raman Spectroscopy, DSC, TGA and other thermal methods, Microscopy and
Solid-state NMR spectroscopy techniques are currently used to characterize these
various crystalline phases. Solving the crystal structure from powder X-ray diffraction
data is slowly becoming a solvable problem.57
To summarize, crystal engineering is an emerging and interdisciplinary subject
of chemistry, physics, biology, materials and pharmaceutical science. This involves
synthesis, crystallography, crystal structure analysis, analysis of all kinds of interactions,
property study, and computation. Study on the molecular recognition events during
nucleation and growth, crystal engineering has acquired control over the internal

30 | Chapter 1
structure and symmetry of crystals and of producing materials with modified chemical
and physical properties. Recent literature reflects the advances in crystal engineering and
its success. This subject is successfully emerged in several exciting new areas of
research, such as catalysis, electronic materials, magnetic sensors, non-linear optics,
nanotechnology, protein-receptor binding, microporous materials, supramolecular
devices, molecular modelling and drug design.
1.13 References 1. (a) J. -M. Lehn, Supramolecular Chemistry, Wiley-VCH, 1995; (b) G. R.
Desiraju, (Eds.), The Crystal as a Supramolecular Entity; Perspectives in
Supramolecular Chemistry, Wiley: Chichester, 1996, Vol. 2; (c) J. W. Steed, J.
L. Atwood, Supramolecular Chemistry, Wiley, Chichester, 2000.
2. (a) W. M. Latimer, W. H. Rodebush, J. Am. Chem. Soc. 1920, 42, 1419; (b) L.
Pauling, The Nature of the Chemical Bond, Cornell University Press, 1939; (c)
L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and
Crystals: An Introduction to Modern Structural Chemistry, Third edition, Ithaca,
NY, Cornell University Press, 1960.
3. (a) G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier,
Amsterdam, 1989; (b) E. Weber (Eds.), Design of Organic Solids, Springer-
Verlag, Berlin, 1998; (c) E. R. T. Tiekink, J. J. Vittal (Eds.), Frontiers in Crystal
Engineering, Wiley, 2006.
4. (a) M. D. Cohen, G. M. J. Schmidt, F. I. Sonntag, J. Chem. Soc. 1964, 2000; (b)
L. Leiserowitz, G. M. J. Schmidt, J. Chem. Soc. A 1969, 2372.
5. J. D. Dunitz, Pure Appl. Chem. 1991, 63, 177.
6. A. I. Kitaigorodskii, Molecular Crystals and Molecules, Academic Press, New
York, 1973.
7. (a) G. R. Desiraju, T. Steiner, The Weak Hydrogen Bond in Structural Chemistry
and Biology, Oxford University Press, Oxford, 1999; (b) G. R. Desiraju, Acc.
Chem. Res. 2002, 35, 565
8. (a) G. R. Desiraju, Angew. Chem. Int. Ed. Engl. 1995, 34, 2311; (b) A. Nangia,
G. R. Desiraju, Top. Curr. Chem. 1998, 198, 57; (c) E.J. Corey, Pure Appl.

31 | Chapter 1
Chem. 1967, 14, 19; (d) E. J. Corey, X. -M. Cheng, The Logic of Chemical
Synthesis, 1989, Wiley, New York.
9. (a) R. D. B. Walsh, M. W. Bradner, S. Fleishman, L. A. Morales, B. Moulton, N.
Rodríguez-Hornedo, M. J. Zaworotko, Chem. Commun. 2003, 186; (b) S. G.
Fleischman, S. S. Kuduva, J. A. McMahon, B. Moulton, R. D. B. Walsh, N.
Rodríguez-Hornedo, M. J. Zaworotko, Cryst. Growth Des. 2003, 3, 909.
10. (a) P. Metrangolo, H. Neukirch, T. Pilati, G. Resnati, Acc. Chem. Res. 2005, 38,
386; (b) E. Corradi, S. V. Meille, M. T. Messina, P. Metrangolo, G. Resnati,
Angew. Chem. Int. Ed. 2000, 39, 1782; (c) N. Ramasubbu, R. Parthasarathy, P.
M. -Rust, J. Am. Chem. Soc. 1986, 108, 4308. (d) S. L. Price, A. J. Stone, J.
Lucas, R. S. Rowland, A. E. Thornley, J. Am. Chem. Soc. 1994, 116, 4910.
11. (a) R. Boese, M. T. Kirchner, W. E. Billups, L. R. Norman, Angew. Chem. Int.
Ed. 2003, 42, 1961; (b) R. Taylor, O. Kennard, J. Am. Chem. Soc. 1982, 104,
5063; (c) Z. S. Derewenda, L. Lee, U. Derewenda, J. Mol. Biol. 1995, 252, 248;
(d) G. R. Desiraju, Acc. Chem. Res. 1996, 29, 441; (e) G. R. Desiraju, Acc.
Chem. Res. 2002, 35, 565; (f) G. R. Desiraju, Chem. Commun. 2005, 2995; (g)
M. C. Wahl, M. Sundaralingam, Trends Biochem. Sci. 1997, 22, 97; (h) Y. M. -
Gutfreund, H. Margalit, R. L. Jernigan, V. B. Zhurkin, J. Mol. Biol. 1998, 277,
1129; (i) M. Nishio, M. Hirota, Y. Umezawa, The C−H/π Interaction, Wiley,
New York, 1988; (j) M. Nishio, CrystEngComm 2003, 7, 130; (k) H. Suezawa,
T. Yoshida, S. Ishihara, Y. Umezawa, M. Nishio, CrystEngComm 2003, 7, 514.
12. (a) M. C. Etter, J. Am. Chem. Soc. 1982, 104, 1095; (b) M. C. Etter, Acc. Chem.
Res. 1990, 23, 120; (c) M. C. Etter, J. Phys. Chem. 1991, 95, 4601.
13. (a) Cambridge Structural Database, CSD, version 5.30, ConQuest 1.11, August
2008 release, May 2009 update; (b) F. H. Allen, Acta Crystallogr. 2002, B58,
380; (c) A. Nangia, CrystEngComm 2002, 4, 93; (d) F. H. Allen, R. Taylor,
Chem. Soc. Rev. 2004, 33, 463; (e) F. H. Allen, W. D. S. Motherwell, Acta.
Crystallogr. 2002, B58, 407.
14. B. Sarma, L. S. Reddy, A. Nangia, Cryst. Growth Des. 2008, 8, 4546.
15. J. Bernstein, R. E. Davis, L. Shimoni, N. L. Chang, Angew. Chem. Int. Ed. Engl.
1995, 34, 1555.

32 | Chapter 1
16. (a) American Heritage Dictionary of the English Language, American Heritage
Publishing Co, and Houghton Mifflin Company, Boston, 1973, pg 319; (b)
http://en.wikipedia.org/wiki/Crystallization
17. (a) S. Parveen, R. J. Davey, G. Dent, R. G. Pritchard, Chem. Commun. 2005,
1531; (b) R. J. Davey, G. Dent, R. K. Mughal, S. Parveen, Cryst. Growth Des.
2006, 6, 1788; (c) R. Banerjee, P. M. Bhatt, M. T. Kirchner, G. R. Desiraju,
Angew. Chem. Int. Ed. 2005, 44, 2515; (d) J. Hulligar, Angew. Chem. Int. Ed.
1994, 33, 143; (e) S. X. M. Boerrigter, F. F. A. Hollander, J. van de Streek, P.
Bennem, H. Meekes, Cryst. Growth Des. 2002, 2, 51; (f) J. Lu, X. -J. Wang, C.
-B. Ching, Cryst. Growth Des. 2003, 3, 83; (g) A. L. Rohl, Current Opinion in
Solid State and Materials Science 2003, 7, 21; (h) H. M. Cuppen, G. M. Day, P.
Verwer, H. Meekes, Cryst. Growth Des. 2004, 4, 1341; (i) R. Hiremath, S. W.
Varney, J. A. Swift, Chem. Commun. 2004, 2676.
18. W. F. Ostwald, Z. Phys. Chem. 1897, 22, 289.
19. (a) J. Bernstein, Polymorphism in Molecular Crystals, Clarendon, Oxford, 2002;
(b) R. Hilfiker, Polymorphism in the Pharmaceutical Industry, Wiley–VCH,
Weinheim, Weinheim, 2006; (c) H. G. Brittan, Polymorphism in Pharmaceutical
Solids, Marcel Dekker, New York, 1999; (d) S. R. Byrn, R. R. Pfeiffer and J. G.
Stowell, Solid-State Chemistry of Drugs, West Lafayette, IN, 1999; (e) T. L.
Threlfall, Analyst 1995, 120, 2435; (f) B. Rodríguez-Spong, C. P. Price, A.
Jayashankar, A. J. Matzger, N. Rodríguez-Hornedo, Adv. Drug. Del. Rev. 2004,
56, 241; (g) J. Bernstein, R. J. Davey, J. -O. Henck, Angew. Chem. Int. Ed. 1999,
38, 3440.
20. (a) G. R. Desiraju, CrystEngComm 2003, 5, 466; (b) J. D. Dunitz,
CrystEngComm 2003, 5, 506; (c) A. D. Bond, CrystEngComm 2007, 9, 833; (d)
J. Z. Schpector, E. R. T. Tiekink, Z. Kristallogr. 2008, 223, 233.
21. (a) A. Nangia, G. R. Desiraju, Chem. Commun. 1998, 605; (b) A. L. Bingham,
D. S. Hughes, M. B. Hursthouse, R. W. Lancaster, S. Tavener, T. L. Threlfall,
Chem. Commun. 2001, 603; (c) A. Nangia, Cryst. Growth Des. 2006, 6, 2; (d) A.
Nangia, Cryst. Growth Des. 2008, 8, 1079; (d) G. R. Desiraju, CrystEngComm
2003, 5, 466; (e) J. D. Dunitz, CrystEngComm 2003, 5, 506; (f) A. D. Bond,
CrystEngComm 2007, 9, 833.

33 | Chapter 1
22. (a) L. Infantes, J. Chisholm, S. Motherwell, CrystEngComm 2003, 5, 480; (b) A.
Gillon, N. Feeder, R. J. Davey, R. Storey, Cryst. Growth Des. 2003, 3, 663; (c)
G. R. Desiraju, J. Chem. Soc. 1991, 6, 426.
23. (a) S. L. Childs, K. I. Hardcastle, Cryst. Growth Des. 2007, 7, 1291; (b) S. L.
Childs, G. P. Stahly, A. Park, Mol. Pharmaceutics 2007, 4, 323.
24. (a) E. Mitscherlich, Abhl. Akad. Berlin 1822-1823, 43; (b) M. J. Buerger, M.
C. Bloom, Z. Kristallogr. 1937, 96, 182; (c) J. Haleblian, W. C McCrone, J.
Pharma. Sci. 1969, 58, 911; (d) J. Berzelius , Jahresbericht 1844, 23, 44; (e) W.
B. Jensen, J. Chem. Educ. 1998, 75, 817; (f) W. C. McCrone, in Physics and
Chemistry of the Organic Solid State, Vol. 2, eds. D. Fox, M. M. Labes and A.
Weissberger, Wiley Interscience, New York, 1965, pp. 725-767; (g) A. Buerger,
in Topics in pharmaceutical science, (Eds.) D. D. Breimer, P. Speiser, Elsevier,
Lausanne, 1983, pp 347-358.
25. (a) V. S. S. Kumar, A. Anthony, A. Nangia, W. T. Robinson, C. K. Broder, R.
Mondal, I. R. Evans, J. A. K. Howard, F. H. Allen, Angew. Chem. Int. Ed. 2002,
41, 3848; (b) N. J. Babu, L. S. Reddy, S. Aitipamula, A. Nangia, Chem Asian J.
2008, 3, 1122; (c) A. Nangia, Acc. Chem. Res. 2008, 41, 595; (d) B. Sarma, S.
Roy, A. Nangia, Chem. Commun. 2006, 4918; (e) B. Sarma, A. Nangia, Acta
Crystallogr. 2008, A64, C449; (f) B. Sarma, A. Nangia (unpublished results).
26. (a) J. A. Bis, M. J. Zaworotko, Cryst. Growth Des. 2005, 5, 1169; (b) S. R.
Chemburkar, J. Bauer, K. Deming, H. Spiwek, K. Patel, J. Morris, R. Henry, S.
Spanton, W. Dziki, W. Porter, J. Quick, P. Bauer, J. Donaubauer, B. A.
Narayanan, M. Soldani, D. Riley, K. McFarland, Org. Process Res. Dev. 2000,
4, 413; (c) J. Bauer, S. Spanton, R. Henry, R. Quick, W. Dziki, W. Porter, J.
Morris, Pharm. Res. 2001, 18, 859; (d) K. Knapman, Mod. Drug. Disc. 2000, 3,
53; (e) S. Datta, D. J. W. Grant, Nat. Rev. Drug. Disc. 2004, 3, 42; (f) Glaxo
Inc. V. Novapharm Ltd. 52F3d 1043, 34 U. S. P. Q. 2d (BNA), 1565 (fed. Cir.
1995).
27. (a) C. M. Reddy, K. A. Padmanabhan, G. R. Desiraju, Cryst. Growth Des. 2006,
6, 2720; (b) C. M. Reddy, S. Basavoju, G. R. Desiraju, Chem. Commun. 2005,
19, 2439; (c) J. Bernstein, Nat. Mater. 2005, 4, 427.

34 | Chapter 1
28. (a) S. L. Morissette, Ö. Almarsson, M. L. Peterson, J. F. Remenar, M. J. Read,
A. V. Lemmo, S. Ellis, M. J. Cima, C. R. Gardner, Adv. Drug. Del. Rev. 2004,
56, 275; (b) A. Linás, J. M. Goodman, Drug. Disc. Today 2008, 13, 198.
29. (a) P. K. Thallapally, R. K. R. Jetti, A. K. Katz, H. L. Carrell, K. Singh, K.
Lahiri, S. Kotha, R. Boese, G. R. Desiraju, Angew. Chem. Int. Ed. 2004, 43,
1149; (b) C. -H. Gu, K. Chatterjee, V. Young Jr, D. J. W. Grant, J. Cryst.
Growth 2002, 235, 471; (c) C. A. Mitchell, L. Yu, M. D. Ward, J. Am. Chem.
Soc. 2001, 123, 10830; (d) X. Sun, B. A. Garetz, A. S. Myerson, Cryst. Growth
Des. 2006, 6, 684; (e) J. L. Hilden, C. E. Reyes, M. J. Kelm, J. S. Tan, J. G.
Stowell, K. R. Morris, Cryst. Growth Des. 2003, 3, 921; (f) G. Di Profio, S.
Tucci, E. Curcio, E. Drioli, Cryst. Growth Des. 2007, 7, 526 (g) M. D. Lang, A.
L. Grzesiak, A. J. Matzger, J. Am. Chem. Soc. 2002, 124, 14834; (h) C. P. Price,
A. L. Grzesiak, A. J. Matzger, J. Am. Chem. Soc. 2005, 127, 5512; (i) A. V.
Trask, N. Shan, W. D. S. Motherwell, W. Jones, S. Feng, R. B. H. Tan, K. J.
Carpenter, Chem. Commun. 2005, 880; (j) A. Bouchard, N. Jovanović, G. W.
Hofland, E. Mendes, D. J. A. Crommelin, W. Jiskoot, G. Witkamp, Cryst.
Growth Des. 2007, 7, 1432; (k) R. Hiremath, J. A. Basile, S. W. Varney, J. A.
Swift, J. Am. Chem. Soc. 2005, 127, 18321; (m) A. Llinás, K. J. Box, J. C.
Burley, R. C. Glen, J. M. Goodman, J. Appl. Crystallogr. 2007, 40, 379.
30. (a) A. Kálmán, L. Fábián, G. Argay, Chem. Commun. 2000, 2255; (b) A.
Nangia, G. R. Desiraju, Acta Crystallogr. 1998, A54, 934; (c) G. R. Desiraju,
Chem. Commun. 1997, 1475; (d) L. Fábián, A. Kálmán, Acta Crystallogr. 1999,
B55, 1099; (e) A. Kálmán, L. Párkányi, G. Argay, Acta Crystallogr. 1993, B49,
1039.
31. (a) http://reference.iucr.org/dictionary/Isomorphous_crystals
(b) http://reference.iucr.org/dictionary/Isostructural_crystals
32. (a) L. Fábián, A. Kálmán, Acta Crystallogr. 2004, B60, 547; (b) A. Anthony, M.
Jaskólski, A. Nangia, G. R. Desiraju, Chem. Commun. 1998, 2537; (c) A.
Anthony, M. Jaskólski, A. Nangia, Acta Crystallogr. 2000, B56, 512.
33. A. I. Kitaigorodskii, Organic Chemical Crsytallography, New York, 1961.

35 | Chapter 1
34. (a) G. Argay, A. Kálmán, B. Ribár, S. Vladimirov, D. Zivanov-Stackic, Acta.
Crystallogr. 1987, C43, 922; (b) A. Kálmán, G. Argay, D. Zivanov-Stackic, S.
Vladimirov, B. Ribár, Acta. Crystallogr. 1992, C48, 812.
35. (a) N. K. Nath, B. K. Saha, A. Nangia, New J. Chem. 2008, 32, 1693; (b) K. F.
Bowes, C. Glidewell, J. N. Low, M. Melguizo, A. Quesada, Acta Crystallogr.
2003, C59, o4; (c) B. Omondi, M. A. fernandes, M. Layh, D. C. Levendis, J. L.
Look, T. S. P. Mkwizu, CrystEngComm 2005, 7, 690; (d) G. R. Desiraju, J. A.
R. P. Sarma, Proc. Ind. Acad. Sci. Chem. Sci. 1986, 96, 599; (e) L. -P. Zhang,
T. C. W. Mak, J. Mol. Struct. 2004, 693, 1; (f) T. Steiner, J. Mol. Struct. 1998,
443, 149; (g) I. Bar, J. Bernstein, Tetrahedron, 1987, 43, 1299; (h) T. A. Olszak,
O. M. Peeters, N. M. Blaton, C. J. de Ranter, Acta Crystallogr. 1994, C50, 761;
(i) L. Prasad, E. J. Gabe, Acta Crystallogr. 1983, C39, 273; (j) S. S. C. Chu, V.
Napoleone, A. I. Jr. Ternay, S. Chang, Acta Crystallogr. 1982, B38, 2508; (k) M.
Chakravarty, P. Kommana, K. C. K. Swamy, Chem. Commun. 2005, 5396; (m)
P. K. Thallapally, K. Chakraborty, H. L. Carrell, S. Kotha, G. R. Desiraju,
Tetrahedron 2000, 56, 6721; (n) P. G. Jones, F. Vancea, CrystEngComm 2003,
5, 303; (o) G. P. Bettinetti, M. R. Caira, M. Sorrenti, L. Catenacci, M. Ghirardi,
L. Fábián, J. Therm. Anal. Cal. 2004, 77, 695; (p) H. Karfunkel, H. Wilts, Z.
Hao, A. Iqbal, J. Mizuguchi, Z. Wu, Acta. Crystallogr. 1999, B55, 1075; (q) N.
Panina, F. J. Leusen, F. B. J. Jassen, P. Verwer, H. Meekes, E. Vleig, G.
Deroover, J. Appl. Cryst. 2007, 40, 105.
36. (a) E. B. Brouwer, K. A. Udachin, G. D. Enright, J. A. Ripmeester, K. J. Ooms,
P. A. Halchuk, Chem. Commun. 2001, 565; (b) P. O. Brown, G. D. Enright, J. A.
Ripmeester, CrystEngComm 2006, 8, 381; (c) B. K. Saha, A. Nangia, Chem.
Commun. 2006, 1825; (d) P. S. Sidhu, G. D. Enright, K. A. Udachin, J. A.
Ripmeester, Chem. Commun. 2005, 2092; (e) E. B. Brouwer, G. D. Enright, K.
A. Udachin, S. Lang, K. J. Ooms, P. A. Halchuk, J. A. Ripmeester, Chem.
Commun. 2003, 1416; (f) J. L. Atwood, L. J. Barbour, G. O. Lloyd, P. K.
Thallapally, Chem. Commun. 2004, 922; (g) G. D. Enright, K. A. Udachin, I. L.
Moudrakovski, J. A. Ripmeester, J. Am. Chem. Soc. 2003, 125, 9896; (h) J. L.
Atwood, L. J. Barbour, A. Jerga, B. L. Schottel, Science 2002, 298, 1000, (i) B.
K. Saha, A. Nangia, CrystEngComm 2006, 8, 440.

36 | Chapter 1
37. (a) J. W. Steed, CrystEngComm 2003, 5, 169; (b) G. R. Desiraju,
CrystEngComm 2007, 9, 91; (c) K. M. Anderson, J. W. Steed, CrystEngComm
2007, 9, 328.
38. (a) S. L. Childs, G. P. Stahly, A. Park, Mol. Pharmaceutics 2007, 4, 323; (b) S.
L. Johnson, K. A. Rumon, J. Phys. Chem. 1965, 69, 74.
39. (a) C. B. Aakeröy, D. J. Salmon, CrystEngComm 2005, 7, 439; (b) B. R.
Bhogala, S. Basavoju, A. Nangia, CrystEngComm 2005, 7, 551; (c) B. R.
Bhogala, S. Basavoju, A. Nangia, Cryst. Growth Des. 2005, 5, 1683; (d) A. V.
Trask, W. D. S. Motherwell, W. Jones, Cryst. Growth Des. 2005, 5, 1013; (e) X.
Gao, T. T. Friščić, L. R. MacGillivray, Angew. Chem. Int. Ed. 2004, 43, 232; (f)
B. R. Bhogala, A. Nangia, Cryst. Growth Des. 2003, 3, 547; (g) C. B. Aakeröy,
A. M. Beatty, B. A. Helfrich, Angew. Chem. Int. Ed. 2001, 40, 3240; (h) B. R.
Bhogala, A. Nangia, New. J. Chem. 2008, 32, 800; (i) P. Vishweshwar, A.
Nangia, V. M. Lynch, J. Org. Chem. 2002, 67, 556; (j) S. Mohamed, D. A.
Tocher, M. Vickers, P. G. Karamertzanis, S. L. Price, Cryst. Growth Des. 2009,
9, 2881.
40. C. Laurence, H. Berthelot, Perspect. Drug. Discovery Des. 2000, 18, 39.
41. Ö. Almarsson, M. J. Zaworotko, Chem. Commun. 2004, 1889.
42. (a) R. D. B. Walsh, M. W. Bradner, S. Fleischman, L. A. Morales, B. Moulton,
N. Rodríguez-Hornedo, M. J. Zaworotko, Chem. Commun. 2003, 186; (b) L.
Childs, L. J. Chyall, J. T. Dunlap, V. N. Smolenskaya, B. C. Stahly, P. G. Stahly,
J. Am. Chem. Soc. 2004, 126, 13335; (c) P. M. Bhatt, N. V. Ravindra, R.
Banerjee, G. R. Desiraju, Chem. Commun. 2005, 1073; (d) R. Banerjee, P. M.
Bhatt, N. V. Ravindra, G. R. Desiraju, Cryst. Growth Des. 2005, 5, 2299; (e) J.
F. Remenar, S. L. Morissette, M. L. Peterson, B. Moulton, J. M. MacPhee, H. R.
Guzman, Ö. Almarsson, J. Am. Chem. Soc. 2003, 125, 8456; (f) M. B. Hickey,
M. L. Peterson, L. A. Scoppettuolo, S. L. Morrisette, A. Vetter, H. Guzmán, J. F.
Remenar, Z. Zhang, M. D. Tawa, S. Haley, M. J. Zaworotko, Ö. Almarsson, Eur.
J. Pharm. Biopharm. 2007, 67, 112; (g) S. L. Morissette, Ö. Almarsson, M. L.
Peterson, J. F. Remenar, M. J. Read, A. V. Lemmo, S. Ellis, M. J. Cima, C. R.
Gardner, Adv. Drug Delivery Rev. 2004, 56, 275; (h) G. Bettinetti, M. R. Caira,
A. Callegari, M. Merli, M. Sorrenti, C. Tadini, J. Pharm. Sci. 2000, 89, 478; (i)

37 | Chapter 1
P. Vishweshwar, J. A. McMahon, M. L. Peterson, M. B. Hickey, T. R. Shattock,
M. J. Zaworotko, Chem. Commun. 2005, 4601; (j) A. V. Trask, W. D. S.
Motherwell, W. Jones, Cryst. Growth Des. 2005, 5, 1013; (k) A. Nangia, N.
Rodríguez-Hornedo, Cryst. Growth Des. 2009, DOI: 10.1021/cg900554h
43. (a) B. R. Sreekanth, P. Vishweshwar, K. Vyas, Chem. Commun. 2007, 2375; (b)
J. A. Bis, P. Vishweshwar, R. A. Middleton, M. J. Zaworotko, Cryst. Growth
Des. 2006, 6, 1048; (c) S. L. Childs, K. I. Hardcastle, Cryst. Growth Des. 2007,
7, 1291; (d) J. A. Bis, P. Vishweshwar, D. Weyna, M. J. Zaworotko, Mol.
Pharma. 2007, 4, 401; (e) W. W. Porter III, S. C. Elie, A. J. Matzger, Cryst.
Growth Des. 2008, 1, 14.
44. (a) C. T. Seto, G. M. Whitesides, J. Am. Chem. Soc. 1990, 112, 6409; (b) N.
Shan, M. J. Zaworotko, Drug. Disc. Today 2008, 13, 440.
45. (a) R. K. Khankari, D. J. W. Grant, Thermochim. Acta. 1995, 61; (b) A. L.
Gillon, N. Feeder, R. J. Davey, R. Storey, Cryst. Growth Des. 2003, 3, 663.
46. (a) R. Custelcean, C. Afloroaei, M. Vlassa, M. Polverejan, Angew. Chem. Int.
Ed. 2000, 39, 3094; (b) R. Ludwig, Angew. Chem. Int. Ed. 2001, 40, 1808; (c) R.
Ludwig, Angew. Chem. Int. Ed. 2003, 42, 258.
47. (a) L. Infantes, J. Chisholm, S. Motherwell, CrystEngComm 2003, 5, 480; (b) L.
Infantes, L. Fábián, W. D. S. Motherwell, CrystEngComm 2007, 9, 65.
48. (a) B. Sarma, A. Nangia, CrystEngComm 2007, 9, 65; (b) B. K. Saha, A. Nangia,
Chem. Commun. 2005, 3024; (c) A. Mukherjee, M. K. Saha, M. Nethaji, A. R.
Chakravarty, Chem. Commun. 2004, 716; (d) F. Kovacs, J. Quine, T. A. Cross,
Proc. Natl. Acad. Sci. USA, 1999, 96, 7910; (e) L. E. Cheruzel, M. S. Pometun,
M. R. Cecil, M. S. Mashuta, R. J. Wittebort, R. M. Buchanan, Angew. Chem. Int.
Ed. 2003, 42, 5452; (f) P. S. Sidhu, K. A. Udachin, J. A. Ripmeester, Chem.
Commun. 2004, 1358; (g) A. Wakahara, T. Ishida. Chem. Lett. 2004, 33, 354.
49. M. Henry, F. Taulelle, T. Loiseau, L. Beitone, G. Féréy, Chem. Eur. J. 2004, 10,
1366.
50. H. Davey, Philos. Trans. R. Soc. Lond. 1811, 101, 155.
51. (a) M. E. Davis, Nature 2002, 417, 813; (b) R. E. Morris, P. S. Wheatley,
Angew. Chem. Int. Ed. 2008, 47, 4966; (c) A. Nangia, Nanoporous Materials:
Science and Engineering, Eds. G. Q. Lu, X. S. Zhao, Imperial College Press,

38 | Chapter 1
London, 2004, pp. 165; (d) L. R. MacGillivray, J. L. Atwood, Angew. Chem. Int.
Ed. 1999, 38, 1018; (e) P. J. Langley, J. Hulliger, Chem. Soc. Rev. 1999, 28, 279;
(f) Y. Aoyama, Top. Curr. Chem. 1998, 198, 131; (g) M. D. Hollingsworth,
Science 2002, 295, 2410; (h) A. Nangia, Curr. Opin. Solid State Mater. Sci.
2001, 5, 115.
52. (a) S. L. James, Chem. Soc. Rev. 2003, 32, 276; (b) S. A. Barnett, N. R.
Champness, Coord. Chem. Rev. 2003, 246, 145; (c) N. W. Ockwig, O. Delgado-
Friedrichs, M. O’Keeffe, O. M. Yaghi, Acc. Chem. Res. 2005, 38, 176; (d) S.
Kitagawa, K. Uemura, Chem. Soc. Rev. 2005, 34, 109.
53. E. Weber, in Comprehensive Supramolecular Chemistry; (Eds.), D. D.
MacNicol, F. Toda, R. Bishop, Pergamon: Oxford, 1996, vol. 6.
54. (a) M. Simard, D. Su, J. D. Wuest, J. Am. Chem. Soc. 1991, 113, 4696; (b) J. D.
Wuest, Chem. Commun. 2005, 5830 and references therein.
55. (a) A. F. Wells, Three-Dimensional Nets and Polyhedra, Wiley, New York,
1977; (b) S. R. Batten, R. Robson, Angew. Chem. Int. Ed. 1998, 37, 1460; (c) L.
Carlucci, G. Ciani, D. M. Proserpio, Coord. Chem. Rev. 2003, 246, 247; (d) L.
Carlucci, G. Ciani, D. M. Proserpio, CrystEngComm 2003, 5, 269; (e) G. R.
Desiraju, Chem. Commun. 1997, 1475; (f) B. Moulton, M. J. Zaworotko, Chem.
Rev. 2001, 101, 1629; (g) M. W. Hosseini, CrystEngComm 2004, 6, 318; (h) R.
E. Melendez, C. V. K. Sharma, M. J. Zaworotko, C. Bauer, R. D. Rogers,
Angew. Chem. Int. Ed. Engl. 1996, 35, 2213; (i) R. K. R. Jetti, P. K. Thallapally,
F. Xue, T. C. W. Mak, A. Nangia, Tetrahedron 2000, 56, 6707; (j) T. C. W.
Mak, F. Xue, J. Am. Chem. Soc. 2000, 122, 9860; (k) R. K. R. Jetti, P. K.
Thallapally, A. Nangia, C. -K. Lam, T. C. W. Mak, Chem. Commun. 2002, 952;
(m) S. Furukawa, M. Obha, S. Kitagawa, Chem. Commun. 2005, 865.
56. (a) S. Aitipamula, A. Nangia, Supramol. Chem. 2005, 17, 17; (b) R. Thakuria, B.
Sarma, A. Nangia, Cryst. Growth Des. 2008, 8, 1471; (c) R. Thakuria, B. Sarma,
A. Nangia, New J. Chem. 2009 (in press).
57. K. D. M. Harris, E. Y. Cheung, Chem. Soc. Rev. 2004, 33, 526.