© 2017 bobby owen garrett
TRANSCRIPT

ENANTIOSELECTIVE HYDROGENATION OF UNFUNCTIONALIZED ALKENES USING
STACKPHOS
By
BOBBY OWEN GARRETT
A THESIS PRESENTED TO THE GRADUATE SCHOOL
OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
OF THE REQUIREMENTS FOR THE DEGREE OF
MASTER OF SCIENCE
UNIVERSITY OF FLORIDA
2017

© 2017 Bobby Owen Garrett

3
ACKNOWLEDGMENTS
I would like to thank first Dr. Aponick for giving me the opportunity to be here; this has
truly been an experience I will always treasure. I’d also like to thank the University of Florida
and the Department of Chemistry for having a great place for people to gather and do science. I
would like to thank my friends along the way that have been with me through the different times
of my life. I hope that even if we don’t see each other every day in the future, that we can still
count on each other as friends. Lastly, I’d like to thank my family. I know the life I’ve lived is a
lot different from the experiences many in my family have had, but they always find a way to
keep me humble and kind.

4
TABLE OF CONTENTS
page
ACKNOWLEDGMENTS ...............................................................................................................3
LIST OF SCHEMES........................................................................................................................5
LIST OF FIGURES .........................................................................................................................6
LIST OF ABBREVIATIONS ..........................................................................................................7
ABSTRACT .....................................................................................................................................9
CHAPTER
1 INTRODUCTION ..................................................................................................................11
Phosphine Experimentation ....................................................................................................11 Atropisomeric Ligand .............................................................................................................12
Iridium Catalysis .....................................................................................................................13 Enantioselective Hydrogenation Using Iridium .....................................................................13 Motivation ...............................................................................................................................15
2 ENANTIOSELECTIVE HYDROGENATION USING STACKPHOS ................................16
Synthesis of Catalyst ...............................................................................................................16
Test Reactions .........................................................................................................................16 Ligand Study ...........................................................................................................................17
1,1-disubstituted Olefins .........................................................................................................18 Synthesis of Substrates ...........................................................................................................19
3 CONCLUSION.......................................................................................................................22
4 EXPERIMENTAL ..................................................................................................................23
1. General Procedure to Synthesize Ligand-Iridium Complex ...............................................24 2. General Procedure for Hydrogenation ................................................................................24
LIST OF REFERENCES ...............................................................................................................44
BIOGRAPHICAL SKETCH .........................................................................................................46

5
LIST OF SCHEMES
Scheme page
1-1 First homogeneous asymmetric hydrogenation. ................................................................11
1-2 Kagan and Knowles rhodium-catalyzed enantioselective hydrogenations. .......................12
1-3 Noyori and Crabtree selective hydrogenations. .................................................................13
1-4 Pfaltz enantioselective hydrogenation of stilbenes. ...........................................................14
1-5 Hydrogenation of minimally functionalized olefins. .........................................................14
2-1 Synthesis of precatalyst for enantioselective hydrogenation. ............................................16
2-2 First attempts of enantioselective hydrogenation. .............................................................16
2-3 Standard hydrogenation conditions, and various ligands used in the reaction. .................18
2-4 Synthesis of test substrates.................................................................................................20

6
LIST OF FIGURES
Figure page
1-1 (S)-StackPhos. ....................................................................................................................15
2-1 First attempts at 1,1-disubstituted hydrogenations. ...........................................................19
2-2 Class of compounds synthesized for hydrogenation. .........................................................20

7
LIST OF ABBREVIATIONS
BARF
BINAP
Tetrakis(3,5-bis(trifluoromethyl)phenyl)borate
2,2'-bis(diphenylphosphino)-1,1'-binaphthyl
br
C
COD
d
DABCO
DCM
DMAP
DMSO
dr
ee
g
h
1H NMR
HPLC
Broad
Celsius
Cyclooctadiene
Doublet
1,4-diazabicyclo[2.2.2]octane
Dichloromethane
4-Dimethylaminopyridine
Dimethylsulfoxide
Diastereomeric excess
Enantiomeric excess
Gram
Hour
Proton nuclear magnetic resonance
High pressure liquid chromatography
HRMS
L-DOPA
High resolution mass spectra
L-3,4-dihydroxyphenylalanine
M
m
MHz
min
mg
mL
Molar
Multiplet
Megahertz
Minute
Milligram
Milliliter

8
mmol
mol
PDC
PHOX
Millimoles
Mole
Pyridinium dichromate
Phosphinooxazolines
psi Pounds per square inch
q
s
t
TBSCl
THF
Quartet
Singlet
Triplet
Tert-butyldimethylsilyl chloride
Tetrahydrofuran
TLC Thin-layer chromatography
TMS Tetramethylsilane

9
Abstract of Thesis Presented to the Graduate School
of the University of Florida in Partial Fulfillment of the
Requirements for the Degree of Master of Science
ENANTIOSELECTIVE HYDROGENATION OF UNFUNCTIONALIZED ALKENES USING
STACKPHOS
By
Bobby Owen Garrett
May 2017
Chair: Aaron Aponick
Major: Chemistry
Ligand-promoted catalysis has played a vital role in the synthesis of complex molecules
due to its ability to accelerate chemical reactions. While there are many classes of ligands used
throughout many fields, P,N-ligands have emerged as highly effective ligands for several
enantioselective transformations. With the development of StackPhos in the Aponick lab in
2013, it was shown that this imidazole-based ligand could achieve high yields and
enantioselectivities in the A3 coupling. Consequently, work has begun both to study the scope of
reactions StackPhos can catalyze enantioselectively and to further study this ligand and
understand its reactivity. To this end, an enantioselective iridium-catalyzed hydrogenation of
unfunctionalized alkenes using StackPhos has been studied. Face selective reduction of alkenes
is one of the most powerful methods to create tertiary and quaternary chiral centers and
synthesize natural products and drug molecules. Initially, a model substrate was studied by
hydrogenating trans-α-methyl stilbene. Once it was confirmed that StackPhos itself was the best
ligand in our study in both reactivity and enantioselectivity, other substrates were tested. Due to
the observed poor reactivity of trans-α-methyl stilbene, it was decided to decrease the steric bulk
around the alkene by moving from a tri-substituted olefin to a 1,1-disubstitued olefin. This
indeed enabled higher conversion of certain substrates, but with others not reacting at all, likely

10
due to electronic effects. While the reactivity of 1,1-disubstituted alkenes was higher, bulky
substrates were still unable to be hydrogenated. Additionally, the enantioselectivity was difficult
to determine by HPLC due to the nonpolar nature of the compounds studied.

11
CHAPTER 1
INTRODUCTION
Enantioselective Hydrogenation: In natural product and drug synthesis, one of the most
vital considerations is how to establish the specified chiral centers. There are many ways to set
chiral centers, and enantioselective hydrogenation is a powerful method used. Hydrogenation of
double bonds is a simple, and straightforward method to produce chiral compounds in an atom-
economical way. High functional group tolerance is observed and often provides desired
compounds with quantitative yields in excellent enantioselectivities.1 While expensive transition
metals such as rhodium and iridium are generally required, they can often be used in less than
1% catalyst loading due to their high turnover rate. The first observed homogenous asymmetric
hydrogenation was pioneered by Knowles at Monsanto in 1968.2 He used an a chiral version of
Wilkinson’s catalyst by replacing triphenylphosphine with chiral phosphine 3 and obtained
minimal selectivity (Scheme 1-1). This was an important preliminary result because the transfer
of ligand chirality to catalyst facial selectivity was achieved in this hydrogenation process.
Scheme 1-1. First homogeneous asymmetric hydrogenation.
Phosphine Experimentation
Significantly higher selectivities were first observed by Kagan in 1971 and Knowles in
1972 with rhodium-catalyzed hydrogenation using ligands 6 and 9 respectively but using a
different substrate (Scheme 1-2).3,4 This moved the field forward by displaying the ability of
rhodium-based catalyst systems to potentially provide high levels of selectivity. In 1975,
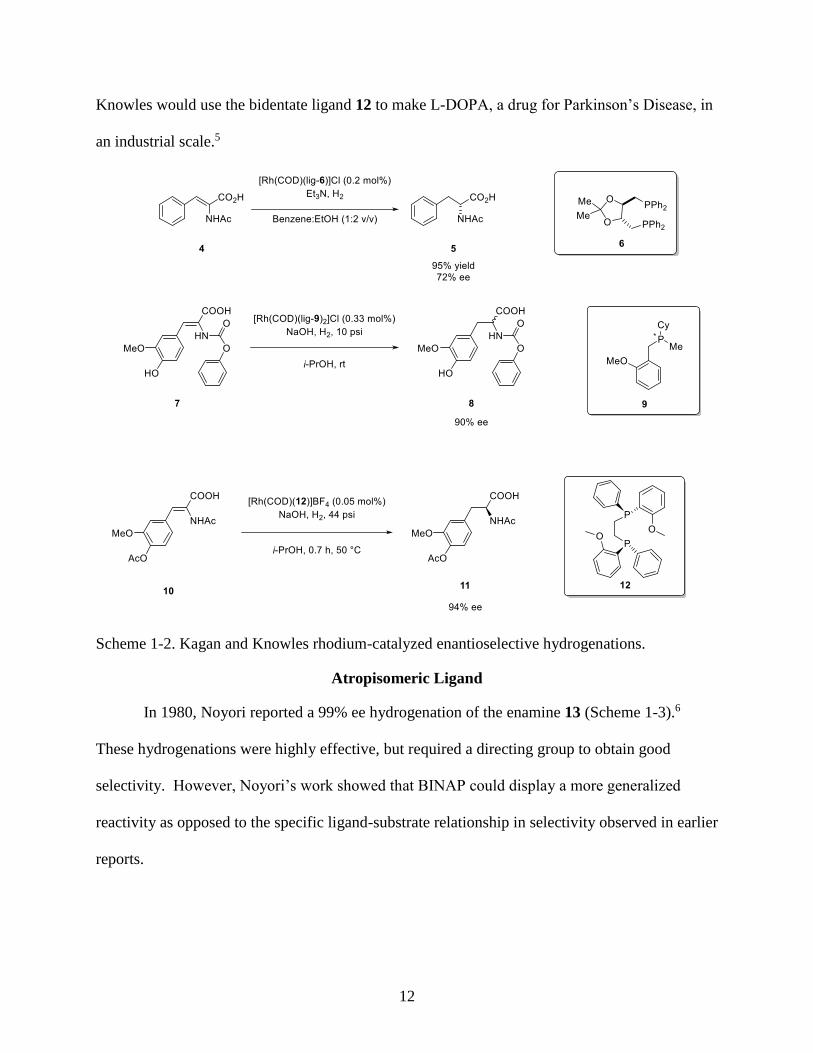
12
Knowles would use the bidentate ligand 12 to make L-DOPA, a drug for Parkinson’s Disease, in
an industrial scale.5
Scheme 1-2. Kagan and Knowles rhodium-catalyzed enantioselective hydrogenations.
Atropisomeric Ligand
In 1980, Noyori reported a 99% ee hydrogenation of the enamine 13 (Scheme 1-3).6
These hydrogenations were highly effective, but required a directing group to obtain good
selectivity. However, Noyori’s work showed that BINAP could display a more generalized
reactivity as opposed to the specific ligand-substrate relationship in selectivity observed in earlier
reports.

13
Iridium Catalysis
Crabtree demonstrated in 1986 that iridium salts could be used as hydrogenation catalysts
as well. He reduced unsaturated menthol 15 with 99% yield and 96:4 dr using what is now
called Crabtree’s catalyst at 1.2 atm.7 However, an alcohol directing group was required in order
to get high selectivity.
Scheme 1-3. Noyori and Crabtree selective hydrogenations.
Enantioselective Hydrogenation Using Iridium
In 1998, the Pfaltz group hydrogenated a series of tri-substituted, unfunctionalized
alkenes. They were able to obtain excellent enantioselectivities by modifying the Crabtree
catalyst using their PHOX-type bidentate P, N ligand instead of tri-cyclohexylphosphine and
pyridine (Scheme 1-4).8

14
Scheme 1-4. Pfaltz enantioselective hydrogenation of stilbenes.
Under their first set of conditions using the iridium-PHOX catalyst with the tetrafluoroborate
anion, only 50% yield was obtained. They found that using a more non-coordinating anion like
BARF dramatically increased the yield to 99% at only 0.3 mol% catalyst loading while retaining
99% ee. When more coordinating anions such as tetrafluoroborate and halides were used, lower
reactivity was observed. Even at 0.05 mol% catalyst loading, they were able to obtain the
product 18 in full conversion, granted at a reduced enantioselectivity. To demonstrate the power
of this method, Pfaltz and coworkers submitted -tocotrienyl acetate 19 to their reaction
conditions using the pyridine-phosphinite ligand 20.9 They selectively obtained 21 as 98% of the
(R,R,R) isomer in quantitative yields (Scheme 1-5).
Scheme 1-5. Hydrogenation of minimally functionalized olefins.

15
Motivation
Because of the aforementioned reactivity of P,N ligands and the high selectivity
observed, hydrogenation using different chiral P,N ligands could afford similar success. Such a
ligand was developed in the Aponick Lab in 2013, namely StackPhos (Figure 1-1.).10 StackPhos
is an axially chiral imidazole-based biaryl P, N-ligand developed in 2013. Many reports exist
using P, N-ligands and iridium catalytic systems, however, most ligands have point chirality or
are axially chiral 6,6-biaryl ligands. Our scaffold has an atropisomeric 6,5-biaryl based system
utilizing aryl-aryl stacking which provides steric and electronic features unique to StackPhos.
This would create a different catalytic environment and could give improved results.
Figure 1-1. (S)-StackPhos.

16
CHAPTER 2
ENANTIOSELECTIVE HYDROGENATION USING STACKPHOS
Synthesis of Catalyst
The first task at hand was to synthesize the hydrogenation catalyst incorporating StackPhos in
order to perform enantioselective hydrogenation (Scheme 2-1).
Scheme 2-1. Synthesis of precatalyst for enantioselective hydrogenation.
Test Reactions
StackPhos was made via the reported procedure.10 The desired metal salt 22 was
obtained quantitatively as a red solid after one hour of stirring (Scheme 2-1). Our test substrate
was trans--methyl stilbene 17 and we subjected it to the reaction conditions for 24 h garnering
only trace amounts of the desired compound 18 (Scheme 2-2).
Scheme 2-2. First attempts of enantioselective hydrogenation.
Decreasing the amount of solvent used from 2 mL to 0.3 mL in order to increase
concentration and heating to 50 °C afforded 37% conversion to the product by 1HNMR. In
Scheme 2-2, only racemic StackPhos had been used in the hydrogenations. From this point
forward, enantiopure Iridium complexes of StackPhos in the range of 95-99% ee were used.

17
With non-racemic catalyst, hydrogenation of 17 gave 33% conversion and 18 in 83% ee. This
result was exciting due to the observed high enantioselectivity, but also because this is the first
example of an elevated temperature reaction utilizing StackPhos. This may indicate that while
the free ligand would racemize at this temperature in the time allotted, StackPhos does not
racemize when complexed to iridium in this way.
Ligand Study
Intrigued by these results, but unsatisfied about heating an enantioselective
transformation for reactivity, changes in reaction conditions were required. The concentration of
the reaction had been increased to as high as possible considering the difficulties in handling the
reaction effectively and dissolving the reagents. We didn’t want to let the reaction go for longer
than 24 h since that was already as long or longer than literature precedents. In further reactions,
we increase the standard pressure of hydrogen to 1000 psi from 725 psi to potentially provide
more reactivity. The other commonly used solvent in these transformations, toluene, was tested
but only provided a 10% conversion. We reduced the catalyst loading, which increased the
conversion to 37%. Having exhausted all other means, modification of the ligand was deemed
necessary and several ligands obtained from other group members were used to test for
reactivity. Me-StackPhos was obtained from Sourabh Mishra in 97% ee as well as racemic
phenanthroline ligand 24. The para-fluoro ligand 23 was obtained from Ji Liu as the racemate.
Benzimidazole ligand 25 was synthesized from an intermediate given by Dr. Cardoso. After
submission to the reaction conditions, Me-StackPhos was the only one besides StackPhos to
show significant yield at 24% and the ee was slightly lower at 67% (Scheme 2-3).

18
Scheme 2-3. Standard hydrogenation conditions, and various ligands used in the reaction.
1,1-disubstituted Olefins
These data showed that reactivity and selectivity of our ligand would probably be
problematic with tri-substituted olefins. As such, it was decided to move to less substituted
olefins since less sterically hindered olefins display increased reactivity. Specifically, the 1,1-
disubstituted alkene moiety was chosen since it is the least hindered alkene that gives a chiral
center upon hydrogenation. Our first attempts to hydrogenate terminal olefins were made with
test substrate -methyl styrene 26 and to our delight, we observed 88% conversion to n-
isopropylbenzene after only 16 h. Hydrogenation of -methyl styrene at 7 M and 0.004%
catalyst loading was attempted and 88% conversion was observed after 24 h. Since n-
isopropylbenzene isn’t chiral and methyl groups aren’t bulky, hydrogenations on two bulkier
compounds were attempted. Both 27 and 28 reacted poorly with minimal product observed for
27 and no product observed for 28 (Figure 2-1).

19
Figure 2-1. First attempts at 1,1-disubstituted hydrogenations.
Synthesis of Substrates
Since sterically bulky groups as both substituents of the alkenes seemed unreasonable as
a test substrate, it was decided to make a set of compounds that had an aryl group on one side
and a small alkyl chain on the other side, namely an ethyl group. This type of substrate was
chosen for several reasons: facial differentiation would occur more easily due to the large
difference in steric bulk between an aryl group and an ethyl group, electronic effects of a
substituent on the aryl ring could potentially be tested to explain differences in reactivity, a chiral
center should enable determination of the facial selectivity of the process. The synthesis of
alkenes of this type would be a simple three-step procedure from either a benzoic acid or a
benzaldehyde. If starting with a benzoic acid, formation of the Weinreb amide11 followed by a
reaction with a Grignard reagent would allow access to the ethyl ketone.12 Starting from the
aldehyde, Grignard addition followed oxidation by pyridinium dichromate13 would yield the
ethyl ketone. The ethyl ketone can undergo the Wittig reaction to obtain the desired alkene
(Scheme 2-4).14

20
Scheme 2-4. Synthesis of test substrates.
The first in this series was the unsubstituted benzene 34. However, problems arose
because the compound was extremely volatile (Figure 2-2). To remedy that, 35 was synthesized
and full conversion was obtained at both 1000 psi and 500 psi. Unfortunately, the product was
too non-polar to study the enantioselectivity by HPLC. The rest of the series was synthesized,
compounds 36-41 using the methods described previously, and complete conversion was
observed for each compound with the exception of the methoxy-substituted benzene.
Figure 2-2. Class of compounds synthesized for hydrogenation.
A potential explanation could be that the electron-donating nature of a methoxy group
inhibits the reactivity of that compound towards hydrogenation. To determine the reactivity of

21
the system is with these compounds, 37 was hydrogenated at 250 psi for 24 h and at atmospheric
pressure for 48 h to give 80% and 78% conversion respectively. However, chiral HPLC
conditions for separation of these compounds could not be determined.

22
CHAPTER 3
CONCLUSION
The enantioselective hydrogenation of 1,1-disubstituted alkenes has been demonstrated
using StackPhos as the chiral ligand. Less bulky, and more electron deficient substrates could be
hydrogenated at lower pressures reliably. Determining the enantioselectivity of this reaction was
difficult due to the non-polar nature of the substrates making them unsuitable to analyze via
traditional chiral HPLC. However, the first example of StackPhos garnering high selectivity at
an elevated temperature has been described, supporting the idea that StackPhos would racemize
more slowly if coordinated strongly as a bidentate ligand to a metal. StackPhos performed far
better than other ligands tested that have varied groups substituted on the imidazole ring.

23
CHAPTER 4
EXPERIMENTAL
General. All reactions were performed under nitrogen atmosphere unless otherwise
stated. Reaction flasks were dried in the oven and anhydrous solvents were transferred via
syringe directly into the flask. Anhydrous dichloromethane (DCM), diethyl ether,
tetrahydrofuran (THF), acetonitrile, and toluene were purified using a mBraun solvent
purification system. Reagents were purchased from Aldrich or Oakwood and used without any
further purification. Analytical thin layer chromatography (TLC) was performed with 250 µm
Silica Gel 60 F254 pre-coated plates (EMD Chemicals Inc.). Flash chromatography utilized
230-400 Mesh 60Å Silica Gel (Whatman Inc.). Proton nuclear magnetic resonance (1HNMR)
spectra were recorded with Varian Unity Inova 500MHz and Varian Mercury 300 MHz
spectrometers. Chemical shifts (δ) are reported in parts per million (ppm) downfield relative to
tetramethylsilane (TMS, 0.0 ppm) or (CDCl3, 7.26 ppm). Coupling constants (J) are reported in
Hz. Multiplicities reported use the following abbreviations: s, singlet; d, doublet; t, triplet; q,
quartet; m, multiplet; br, broad. Carbon-13 nuclear magnetic resonance (13C NMR) spectra were
obtained using a Varian Unity Mercury 300 spectrometer at 75 MHz. Chemical shifts are
reported in ppm relative to the carbon resonance of CDCl3 (77.00 ppm). Infrared spectra were
obtained on a Bruker Vector 22 IR spectrometer at 4.0 cm-1 resolution and are reported in
wavenumbers. High resolution mass spectra (HRMS) were obtained by Mass Spectrometry Core
Laboratory of University of Florida, and are reported as m/e (relative ratio). Accurate masses are
reported for the molecular ion (M+) or a suitable fragment ion.

24
1. General Procedure to Synthesize Ligand-Iridium Complex
To a flask of NaBARF (20 mg, 0.0225 mmol) and [Ir(COD)Cl]2 (50 mg, 0.0075 mmol) was
added a solution of ligand (0.015 mmol) in dichloromethane (0.5 mL). The resulting orange
solution was stirred at room temperature for 1 h. The solution was filtered through a plug of
silica and concentrated to give the desired compound as a red or orange crystalline solid.8
2. General Procedure for Hydrogenation
To a test tube (13x100 mm) was added an iridium catalyst (0.00107 mmol), the precursor alkene
(0.330 mmol), and dichloromethane (0.3 mL) to form a red solution. The test tube was placed
inside a Parr bomb, sealed, vented with hydrogen, then pressurized to 1000 psi and stirred for 24
h. The reaction mixture was concentrated by carefully blowing a stream of nitrogen into the test
tube, dissolved in hexanes, filtered through a plug of silica, and concentrated to give the
compound as a colorless or pale yellow oil.8

25
Prepared according to a previous report in the Aponick group.10 Spectroscopic data matches the
reported values.
Prepared by a similar procedure.10 To a solution of 1-(1H-benzo[d]imidazol-2-yl)naphthalen-2-ol
provided by Dr. Cardoso (332 mg, 1.28 mmol) and triethylamine (0.178 mL, 1.28 mmol) in
DCM (10 mL) was added TBSCl (192 mg, 1.28 mmol) at room temperature for 1.5 h and
quenched with water (2 mL). The crude compound was extracted with DCM (3 x 1 mL), dried
over MgSO4 and the solvent removed under pressure. The solid residue was purified by flash
chromatography (5-20% ethyl acetate/hexanes) to yield 356 mg (74%) of 42 as a white solid. 1H
NMR (300 MHz, CDCl3) δ 8.62 (d, J= 6 Hz, 1H), 7.95 (m, 2H), 7.76 (br, 2H), 7.51 (quint, J=
6Hz, 2H), 7.38 (m, 2H), 7.26 (d, J= 9 Hz, 1H), 0.99 (s, 6H), 0.95 (s, 9H).

26
Prepared by a similar procedure.10 Compound 42 (356 mg, 0.953 mmol) dissolved in THF (5
mL) was added dropwise to a suspension of sodium hydride (24 mg, 1.0 mmol) in THF (5 mL) at
-78 °C. After ten minutes of stirring, pentafluorobenzyl bromide (0.144 mL, 0.953 mmol) was
added dropwise to the solution. The reaction was allowed to warm to room temperature and stir
for 18 h at which point the solution was cooled to 0 °C and water was added. The organic phase
was separated and the aqueous phase extracted with ethyl acetate (3 x 1 mL). The combined
organic layers were dried over MgSO4 and concentrated under reduced pressure. The residue
was purified by flash chromatography (5-20% ethyl acetate/hexanes) to yield 122 mg (23%) of
43 as an oil. 1H NMR (300 MHz, CDCl3) δ 7.97 (d, J= 6 Hz, 2H), 7.82 (d, J= 3 Hz, 1H), 7.50
(m, 1H), 7.38 (m, 5H), 7.19 (d, J= 6 Hz, 2H), 5.35 (q, J= 6 Hz, 1H), 1.59 (s, 6H), 0.78 (s, 9H).
Prepared by a similar procedure.10 To a solution of 43 (582 mg, 1.05 mmol) in methanol (30
mL) was added potassium carbonate (290 mg, 2.10 mmol) at room temperature and stirred for 30
min. To the solid was added ethyl acetate and water, then the organic layer was removed and the
aqueous layer extracted with ethyl acetate. After drying with magnesium sulfate and

27
concentrating under reduced pressure, the deprotected alcohol was obtained as a brown solid and
used as crude in the next step.
To a solution of the compound and DMAP (12.8 mg, 0.105 mmol) in dichloromethane (30 mL)
was added triethylamine (0.146 mL, 1.05 mmol) and triflic anhydride (0.176 mL, 1.05 mmol) at
room temperature for 6 h. The reaction was then concentrated and purified by flash
chromatography (5-10% ethyl acetate/hexanes) to yield 420 mg (70%) of 44. 1H NMR (300
MHz, CDCl3) δ 8.26 (d, J= 6 Hz, 1H), 8.08 (d, J= 6 Hz, 2H), 7.70 (m, 3H), 7.58 (m, 4H), 5.43
(q, J= 12 Hz, 6H).
Prepared by a similar procedure.10 To nickel(II)-1,2-bis(diphenylphosphino)ethane dichloride
(38.5 mg, 0.073 mmol) dissolved in dimethylformamide (4 mL), diphenylphosphine (0.256 mL,
1.47 mmol) was added to the solution and stirred at 130°C for 30 min. DABCO (329 mg, 2.93
mmol) and 44 (420 mg, 0.73 mmol) were then dissolved in dimethylformamide (3 mL), added to
the dark red solution, and stirred at 130°C for 14 h. The dimethylformamide was then removed
via distillation at reduced pressure and the compound was purified via flash chromatography (5-
20% ethyl acetate/hexanes gradient) to yield 177 mg (40%) 25 as a pale yellow solid. 1H NMR
(300 MHz, CDCl3) δ 8.01 (d, J= 6 Hz, 1H), 7.93 (d, J= 6 Hz, 2H), 7.66 (d, J= 3 Hz, 1H), 7.55
(m, 9H), 7.31 (m, 6H), 7.03 (d, J= 6Hz, 1H), 5.30 (q, J= 6 Hz, 2H).

28
Prepared according to general procedure 1 to yield 20.4 mg (73%) of the desired compound. 1H
NMR (500 MHz, CDCl3) δ 8.06 (m, 2H), 7.92 (d, J= 10 Hz, 1H), 7.74 (m, 14H), 7.52 (m, 11H),
7.33 (t J= 10 Hz, 1H), 7.05 (t, J= 10 Hz, 2H), 5.18 (br, 1H), 4.90 (d, J= 20 Hz, 1H), 4.50 (d, J=
30 Hz, 1H), 4.10 (br, 1H), 3.31 (br, 1H), 2.81 (br, 1H), 2.42 (br, 1H), 2.11(m, 1H), 2.04 (br, 2H),
1.76 (br, 1H), 0.87 (br, 1H); HRMS (ESI) calcd for C52H40F5IrN2P [M-BARF]+ 1011.2479,
found 1011.2512.
Prepared according to general procedure 1 to yield 14.2 mg (99%) of the desired compound. 1H
NMR (500 MHz, CDCl3) δ 8.09 (dd J= 10, 5 Hz, 2H), 7.88 (d J= 5 Hz, 1H), 7.73 (m, 17H), 7.55
(m, 9H), 7.18 (t, J=10 Hz, 2H), 7.03 (m, 4H), 5.19 (br, 1H), 4.86 (d, J= 20 Hz, 1H), 4.59 (d, J=
20 Hz, 1H), 4.07 (br, 1H), 3.19 (m, 1H), 2.85 (br, 1H), 2.40 (br, 1H), 2.09 (m, 1H), 2.04 (m, 3H),
1.89 (br, 1H), 0.87 (m, 1H).

29
Prepared according to general procedure 1 but at half the molarity to yield 12 mg (91%) of the
desired compound.
Prepared according to general procedure 1 to yield 7.7 mg (55%) of the desired compound. 1H
NMR (500 MHz, CDCl3) δ 8.79 (t J= 5, Hz, 2H), 8.18 (d J= 10 Hz, 1H), 8.02 (m, 4H), 7.92 (t J=
5 Hz, 1H), 7.88 (t, J= 10 Hz, 1H), 7.77 (m, 6H), 7.63 (m, 1H), 7.56 (m, 7H), 7.18 (m, 2H), 7.04
(br, 2H), 5.61 (d, J= 20 Hz, 1H), 5.01 (br, 1H), 4.92 (d, J= 20 Hz, 1H), 4.60 (br, 1H), 4.18 (m,
1H), 3.21 (br, 1H), 2.98 (m, 1H), 2.43 (m, 1H), 1.97 (m, 2H), 1.61 (br, 1H), 1.42 (br, 1H), 0.89
(br, 1H).

30
Prepared according to general procedure 1 but at four times the molarity to yield 50.1 mg (92%)
of the desired compound. 1H NMR (500 MHz, CDCl3) δ 8.31 (d J= 10 Hz, 2H), 8.18(d J= 10
Hz, 1H), 7.82 (m, 13H), 7.62 (m, 9H), 7.33 (t, J= 5 Hz, 1H), 7.21 (t, J= 5 Hz, 2H), 7.05 (t, J= 5
Hz, 2H), 5.55 (br, 1H), 5.23 (t, J= 5 Hz, 1H), 5.01 (q, J= 20 Hz, 2H), 4.21 (br, 1H), 3.10 (br,
1H), 2.73 (d, J= 20 Hz, 1H), 2.01 (br, 1H), 1.82 (br, 1H), 1.58 (br, 1H), 0.99 (br, 1H).
Prepared according to general procedure 2. 1H NMR (300 MHz, CDCl3) δ 7.32 (m, 10H), 3.00
(m, 2H), 2.80 (m, 1H), 1.30 (d J= 6 Hz, 3H).
To methyltriphenylphosphonium bromide (2.036 g, 5.67 mmol) in diethyl ether (20 mL) at 0°C
was added n-butyl lithium solution in hexanes (2.27 mL, 5.67 mmol) and stirred until the
solution turned orange (half an hour). To the resulting orange solution, ketone (0.503 mL, 3.78
mmol) was slowly added and solids immediately formed. The reaction was then stirred at room

31
temperature for 16 h. The reaction mixture was concentrated and a column was ran with 100%
hexanes to yield 208 mg (42%) as a pale yellow oil. Spectra matches previously reported data.17
1H NMR (300 MHz, CDCl3) δ 7.52 (m, 1H), 7.38 (m, 4H), 5.39 (d, J= 3 Hz, 1H), 5.18 (d, J= 3
Hz, 1H), 2.63 (t, J= 6 Hz, 3H), 1.22 (q, J= 6 Hz, 2H).
To methyltriphenylphosphonium bromide (2.036 g, 5.67 mmol) in diethyl ether (20 mL) at 0°C
was added n-butyl lithium solution in hexanes (2.27 mL, 5.67 mmol) and stirred until the
solution turned orange (half an hour). To the resulting orange solution, ketone (712 mg, 3.78
mmol) was slowly added and solids immediately formed. The reaction was then stirred at room
temperature for 16 h. The reaction mixture was concentrated and a column was ran with 100%
hexanes to yield the compound as a pale yellow oil. Spectra matches previously reported data.15
1H NMR (300 MHz, CDCl3) δ 7.57 (5H), 5.42 (s, 1H), 5.26 (s, 1H), 2.68 (t, J= 9 Hz, 1H), 2.02
(m, 6H), 1.55 (m, 6H).
To methyltriphenylphosphonium bromide (2.46 mg, 6.9 mmol) in diethyl ether (23 mL) at 0°C
was added n-butyl lithium solution in hexanes (4.31 mL, 6.9 mmol) and stirred until the solution
turned orange (half an hour). To the resulting orange solution, ketone (752 mg, 4.6 mmol) was
slowly added and solids immediately formed. The reaction was then stirred at room temperature
for 16 h. The reaction mixture was concentrated and a column was ran with 100% hexanes to

32
yield 403 mg (54%) as a yellow oil. Spectra matches previously reported data.17 1H NMR (300
MHz, CDCl3) δ 7.39 (d, J=6 Hz, 2H), 6.87 (d, J=6 Hz, 2H), 5.22 (s, 1H), 4.99 (s, 1H), 3.82 (s,
3H), 2.52 (q, J= 6 Hz, 2H), 1.11 (t, J= 6 Hz, 3H).
To methyltriphenylphosphonium bromide (3.24 g, 9.1 mmol) in benzene (45 mL) at 0°C was
added n-butyl lithium solution in hexanes (5.7 mL, 9.1 mmol) and stirred until the solution
turned orange (half an hour). To the resulting orange solution, ketone (1.22 g, 6.1 mmol) was
slowly added and solids immediately formed. The reaction was then stirred at room temperature
for 16 h. The reaction mixture was concentrated and a column was ran with 100% hexanes to
yield 284 mg (23%) as a pale yellow oil. Spectra matches previously reported data.17 1H NMR
(300 MHz, CDCl3) δ 7.61 (d, J=6 Hz, 2H), 7.53 (d, J=6 Hz, 2H), 5.36 (s, 1H), 5.18 (s, 1H), 2.55
(q, J= 6 Hz, 2H), 1.3 (t, J= 6 Hz, 3H).
To methyltriphenylphosphonium bromide (1.39 g, 3.9 mmol) in diethyl ether (15 mL) at 0°C was
added n-butyl lithium solution in hexanes (2.44 mL, 3.9 mmol) and stirred until the solution
turned orange (half an hour). To the resulting orange solution, ketone (434 mg, 2.6 mmol) was
slowly added and solids immediately formed. The reaction was then stirred at room temperature
for 16 h. The reaction mixture was concentrated and a column was ran with 100% hexanes to

33
yield 95.5 mg (22%) as a yellow oil. Spectra matches previously reported data.19 1H NMR (300
MHz, CDCl3) δ 7.42 (m, 4H), 5.39 (s, 1H), 5.19 (s, 1H), 2.60 (q, J= 6 Hz, 2H), 1.21 (t, J= 6 Hz,
3H).
To methyltriphenylphosphonium bromide (1.39 g, 3.9 mmol) in diethyl ether (15 mL) at 0°C was
added n-butyl lithium solution in hexanes (2.44 mL, 3.9 mmol) and stirred until the solution
turned orange (half an hour). To the resulting orange solution, ketone (495 mg, 2.6 mmol) was
slowly added and solids immediately formed. The reaction was then stirred at room temperature
for 16 h. The reaction mixture was concentrated and a column was ran with 100% hexanes to
yield 181.5 mg (37%) as a yellow oil. Spectra matches previously reported data.18 1H NMR (300
MHz, CDCl3) δ 7.26 (m, 4H), 5.25 (s, 1H), 5.01 (s, 1H), 2.48 (q, J= 6 Hz, 2H), 1.33 (s, 9H), 1.10
(t, J= 6 Hz, 3H).
To methyltriphenylphosphonium bromide (1.18 g, 3.3 mmol) in diethyl ether (15 mL) at 0°C was
added n-butyl lithium solution in hexanes (2.06 mL, 3. 3mmol) and stirred until the solution
turned orange (half an hour). To the resulting orange solution, ketone (333 mg, 2.2 mmol) was
slowly added and solids immediately formed. The reaction was then stirred at room temperature
for 16 h. The reaction mixture was concentrated and a column was ran with 100% hexanes to

34
yield 53.9 mg (16%) as a yellow oil. Spectra matches previously reported data.20 1H NMR (300
MHz, CDCl3) δ 7.42 (m, 4H), 5.39 (s, 1H), 5.19 (s, 1H), 2.60 (q, J= 6 Hz, 2H), 1.21 (t, J= 6 Hz,
3H).
To methyltriphenylphosphonium bromide (893 mg, 2.5 mmol) in diethyl ether (10 mL) at 0°C
was added n-butyl lithium solution in hexanes (1.56 mL, 2.5 mmol) and stirred until the solution
turned orange (half an hour). To the resulting orange solution, ketone (355 mg, 1.7 mmol) was
slowly added and solids immediately formed. The reaction was then stirred at room temperature
for 16 h. The reaction mixture was concentrated and a column was ran with 100% hexanes to
yield 33.5 mg (9%) as a yellow oil. Spectra matches previously reported data.19 1H NMR (300
MHz, CDCl3) δ 7.42 (m, 4H), 5.39 (s, 1H), 5.19 (s, 1H), 2.60 (q, J= 6 Hz, 2H), 1.21 (t, J= 6 Hz,
3H).
Prepared according to general procedure 2. Spectra matches previously reported data.17 1H NMR
(300 MHz, CDCl3) δ 7.42 (m, 4H), 7.32 (m, 6H), 2.73 (m, 1H), 1.75 (m, 2H), 1.38 (d, J= 6 Hz,
3H), 0.98 (t, J= 6 Hz, 3H).

35
Prepared according to general procedure 2. Spectra matches previously reported data.21 1H NMR
(300 MHz, CDCl3) δ 7.82 (m, 3H), 7.62 (m, 1H), 7.43 (m, 3H), 2.80 (m, 1H), 1.75 (m, 2H), 1.38
(d, J= 6 Hz, 3H), 0.87 (t, J= 6 Hz, 3H).
To methyltriphenylphosphonium bromide (2.036 g, 5.67 mmol) in diethyl ether (20 mL) at 0°C
was added n-butyl lithium solution in hexanes (2.27 mL, 5.67 mmol) and stirred until the
solution turned orange (half an hour). To the resulting orange solution, ketone (666 mg, 3.78
mmol) was slowly added and solids immediately formed. The reaction was then stirred at room
temperature for 16 h. The reaction mixture was concentrated and a column was ran with 100%
hexanes to yield 478 mg (73%) as a yellow oil. Spectra matches previously reported data.16 1H
NMR (300 MHz, CDCl3) δ 7.68 (d, J= 6 Hz, 2H), 7.01 (d, J= 6 Hz, 2H), 5.36 (s, 1H), 5.00 (s,
1H), 3.97 (s, 3H), 1.77 (m, 1H), 1.08 (m, 4H).
Prepared according to general procedure 2. Spectra matches previously reported data.17 1H NMR
(300 MHz, CDCl3) δ 7.58 (d, J= 6 Hz, 2H), 7.35 (d, J= 6 Hz, 2H), 2.68 (m, 1H), 1.63 (m, 2H),
1.28 (d, J= 6 Hz, 3H), 0.86 (t, J= 6 Hz, 3H).

36
Prepared according to general procedure 2. Spectra matches previously reported data.19 1H NMR
(300 MHz, CDCl3) δ 7.28 (d, J= 6 Hz, 2H), 7.16 (d, J= 6 Hz, 2H), 2.60 (m, 1H), 1.61 (m, 2H),
1.28 (d, J= 6 Hz, 3H), 0.86 (t, J= 6 Hz, 3H).
Prepared according to general procedure 2. Spectra matches previously reported data.22 1H NMR
(300 MHz, CDCl3) δ 7.39 (d, J= 6 Hz, 2H), 7.18 (d, J= 6 Hz, 2H), 2.64 (m, 1H), 1.65 (m, 2H),
1.41 (s, 9H), 1.32 (d, J= 6 Hz, 3H), 0.92 (t, J= 6 Hz, 3H).
Prepared according to general procedure 2. Spectra matches previously reported data.19 1H NMR
(300 MHz, CDCl3) δ 7.41 (d, J= 6 Hz, 2H), 7.07 (d, J= 6 Hz, 2H), 2.58 (m, 1H), 1.59 (m, 2H),
1.22 (d, J= 6 Hz, 3H), 0.83 (t, J= 6 Hz, 3H).

37
To methyltriphenylphosphonium bromide (478 g, 1.34 mmol) in diethyl ether (20 mL) at 0°C
was added n-butyl lithium solution in hexanes (0.797 mL, 1.34 mmol) and stirred until the
solution turned orange (half an hour). To the resulting orange solution, ketone (164.3 mg, 0.89
mmol) was slowly added and solids immediately formed. The reaction was then stirred at room
temperature for 16 h. The reaction mixture was concentrated and a column was ran with 100%
hexanes to yield 69.6 mg (43%) as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 7.99 (m, 4H),
7.79 (d J= 3 Hz, 1H) 7.63 (m, 2H), 5.62 (s, 1H), 5.38 (s, 1H), 2.84 (q, J= 6 Hz, 2H), 1.38 (t, J= 6
Hz, 3H).
Prepared according to general procedure 2. 1H NMR (300 MHz, CDCl3) δ 7.59 (m, 2H), 7.29
(m, 2H), 6.68 (m, 1H), 1.63 (dd, J= 6 Hz, 2H), 1.28 (d, J= 6 Hz, 3H), 0.87 (t, J= 6 Hz, 3H).
To aldehyde (0.89 mL, 7.3 mmol) in diethyl ether (35 mL) in a flamed dried flask at 0° C was
added 3M ethylmagnesium bromide in THF (3.67 mL, 11 mmol). The resulting pale yellow
solution was then allowed to stir at room temperature for 2 hours. The reaction was quenched at
0°C with 1M hydrochloric acid and was extracted with ethyl acetate. The resulting organic layer

38
was dried with brine and sodium sulfate. Upon concentration under reduced pressure, 1.11 g
(100%) of a colorless oil was obtained as the product. 1H NMR (300 MHz, CDCl3) δ 7.39 (d,
J=6 Hz, 2H), 6.99 (d, J=6 Hz, 2H), 4.63 (br, 1H), 3.92 (s, 3H), 2.53 (br, 1H), 1.88 (m, 2H), 1.02
(t, J= 6 Hz, 3H).
To a solution of oxalyl chloride in dichloromethane at -78°C was added DMSO dropwise. After
1 h the alcohol was added dropwise. After 30 min triethylamine was added dropwise and the
reaction was allowed to warm to room temperature and stirred overnight. The reaction
concentrated under reduced pressure and flash chromatography (10% ethyl acetate/hexanes) gave
751.7 mg (63%) of the desired product. 1H NMR (300 MHz, CDCl3) δ 7.98 (d, J=6 Hz, 2H),
6.94 (d, J=6 Hz, 2H), 3.85 (s, 3H), 2.96 (q, J= 6 Hz, 2H), 1.22 (t, J= 6 Hz, 3H).
To alcohol (1.16 g, 5.7 mmol) in dichloromethane (30 mL) was added pyridinium dichromate
(3.22 g, 8.6 mmol) and stirred for 16 h at room temperature. The resulting brown solution was
filtered over celite and concentrated under reduced pressure to yield 627.2 mg (54%) as a pale
yellow oil. 1H NMR (300 MHz, CDCl3) δ 8.08 (d, J=6 Hz, 2H), 7.75 (d, J=6 Hz, 2H), 3.03 (q,
J= 6 Hz, 2H), 1.24 (t, J= 6 Hz, 3H).

39
To alcohol (765 g, 4.5 mmol) in dichloromethane (30 mL) was added pyridinium dichromate
(2.53 g, 6.7 mmol) and stirred for 16 h at room temperature. The resulting brown solution was
filtered over celite and concentrated under reduced pressure to yield 435 mg (57%) as a pale
yellow oil. 1H NMR (300 MHz, CDCl3) δ 8.03 (d, J=6 Hz, 2H), 7.58 (d, J=6 Hz, 2H), 3.08 (q,
J= 6 Hz, 2H), 1.81 (t, J= 6 Hz, 3H).
To alcohol (898 g, 4.7 mmol) in dichloromethane (30 mL) was added pyridinium dichromate
(2.635 g, 7.0 mmol) and stirred for 16 h at room temperature. The resulting brown solution was
filtered over celite and concentrated under reduced pressure to yield 495 mg (55%) as a pale
yellow oil. 1H NMR (300 MHz, CDCl3) δ 7.95 (d, J=6 Hz, 2H), 7.49 (d, J=6 Hz, 2H), 2.99 (q,
J= 6 Hz, 2H), 1.79 (s, 9H), 1.39 (t, J= 6 Hz, 3H).
To alcohol (835 g, 5.4 mmol) in dichloromethane (30 mL) was added pyridinium dichromate
(3.06 g, 8.1 mmol) and stirred for 16 h at room temperature. The resulting brown solution was
filtered over celite and concentrated under reduced pressure to yield 333 mg (41%) as a pale

40
yellow oil. 1H NMR (300 MHz, CDCl3) δ 8.01 (t, J=6 Hz, 2H), 7.15 (t, J=6 Hz, 2H), 2.98 (q, J=
6 Hz, 2H), 1.22 (t, J= 6 Hz, 3H).
To Weinreb amide (409 mg, 1.6 mmol) in diethyl ether (10 mL) in a flamed dried flask at 0° C
was added 3M ethylmagnesium bromide in THF (1.1 mL, 3.3 mmol). The resulting pale yellow
solution was then allowed to stir at room temperature for 2 hours. The reaction was quenched at
0°C with 1M hydrochloric acid and was extracted with ethyl acetate. The resulting organic layer
was dried with brine and sodium sulfate. Upon concentration under reduced pressure, 355 mg
(100%) of the desired compound a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.95 (d J= 6 Hz,
2H), 7.73 (d J= 6 Hz, 2H), 3.09 (q, J= 6 Hz, 2H), 1.36 (t, J= 6 Hz, 3H).
To aldehyde (801 mg, 5.7 mmol) in diethyl ether (30 mL) in a flamed dried flask at 0° C was
added 3M ethylmagnesium bromide in THF (3.8 mL, 11.4 mmol). The resulting pale yellow
solution was then allowed to stir at room temperature for 2 hours. The reaction was quenched at
0°C with 1M hydrochloric acid and was extracted with ethyl acetate. The resulting organic layer
was dried with brine and sodium sulfate. Upon concentration under reduced pressure, 765 mg
(79%) of a colorless oil was obtained as the product. 1H NMR (300 MHz, CDCl3) δ 7.64 (d, J=6

41
Hz, 2H), 7.47 (d, J=6 Hz, 2H), 4.69 (br, 1H), 1.80 (q, J= 6 Hz, 2H), 1.58 (br, 1H), 0.98 (t, J= 6
Hz, 3H).
To Weinreb amide (456 mg, 2.1 mmol) in diethyl ether (3 mL) in a flamed dried flask at 0° C
was added 3M ethylmagnesium bromide in THF (2.24 mL, 6.7 mmol). The resulting pale
yellow solution was then allowed to stir at room temperature for 2 hours. The reaction was
quenched at 0°C with 1M hydrochloric acid and was extracted with ethyl acetate. The resulting
organic layer was dried with brine and sodium sulfate. Upon concentration under reduced
pressure, 164 mg (42%) of the desired compound a colorless oil. 1H NMR (300 MHz, CDCl3) δ
8.59 (s, 1H), 8.17 (d J= 3 Hz, 1H) 8.09 (d J= 3 Hz, 1H), 7.97 (m, 2H), 7.67 (m, 2H), 3.24 (q, J=
6 Hz, 2H), 1.02 (t, J= 6 Hz, 3H).
To aldehyde (0.78 mL, 5.7 mmol) in diethyl ether (30 mL) in a flamed dried flask at 0° C was
added 3M ethylmagnesium bromide in THF (3.8 mL, 11.4 mmol). The resulting pale yellow
solution was then allowed to stir at room temperature for 2 hours. The reaction was quenched at
0°C with 1M hydrochloric acid and was extracted with ethyl acetate. The resulting organic layer
was dried with brine and sodium sulfate. Upon concentration under reduced pressure, 223 mg

42
(11%) of a colorless oil was obtained as the product. 1H NMR (300 MHz, CDCl3) δ 7.62 (d J= 6
Hz, 2H), 7.47 (t, J=6 Hz, 2H), 4.69 (br, 1H), 1.82 (br, 3H), 0.96 (t, J= 6 Hz, 3H).
To a solution of carboxylic acid (1.0 g, 4.97 mmol) in benzene (20 ml) was added thionyl
chloride (0.86 mL, 11.71 mmol) and refluxed for 1 h. The mixture was concentrated under
reduced pressure and dissolved in dichloromethane (20 mL). Then, N,O-
Dimethylhydroxylamine hydrochloride (530 mg, 5.48 mmol) and pyridine (0.89 mL, 11.05
mmol) were added to the reaction. After 14 h the solution was quenched with saturated sodium
carbonate solution and washed with water and brine before being dried with sodium sulfate,
filtered, and concentrated under reduced pressure to yield 409 mg (32%) Weinreb amide as a
colorless oil. 1H NMR (300 MHz, CDCl3) δ 8.00 (d J= 6 Hz, 2H), 7.73 (t, J= 6 Hz, 2H), 3.53 (s,
3H), 3.37 (s, 3H).
To aldehyde (0.953 mL, 5.7 mmol) in diethyl ether (30 mL) in a flamed dried flask at 0° C was
added 3M ethylmagnesium bromide in THF (3.8 mL, 11.4 mmol). The resulting pale yellow
solution was then allowed to stir at room temperature for 2 hours. The reaction was quenched at
0°C with 1M hydrochloric acid and was extracted with ethyl acetate. The resulting organic layer
was dried with brine and sodium sulfate. Upon concentration under reduced pressure, 898 mg

43
(82%) of a colorless oil was obtained as the product. 1H NMR (300 MHz, CDCl3) δ 7.48 (d, J=6
Hz, 2H), 7.39 (d, J=6 Hz, 2H), 4.68 (t, J= 3 Hz, 1H), 1.88 (br, 3H), 1.03 (t, J= 6 Hz, 3H).
To aldehyde (0.611 mL, 5.7 mmol) in diethyl ether (30 mL) in a flamed dried flask at 0° C was
added 3M ethylmagnesium bromide in THF (3.8 mL, 11.4 mmol). The resulting pale yellow
solution was then allowed to stir at room temperature for 2 hours. The reaction was quenched at
0°C with 1M hydrochloric acid and was extracted with ethyl acetate. The resulting organic layer
was dried with brine and sodium sulfate. Upon concentration under reduced pressure, 836 mg
(95%) of a colorless oil was obtained as the product. 1H NMR (300 MHz, CDCl3) δ 7.43 (m,
2H), 7.15 (t, J=6 Hz, 2H), 4.70 (br, 1H), 2.90 (br, 3H), 1.02 (t, J= 6 Hz, 3H).
To a solution of carboxylic acid (1.032 g, 6.0 mmol), N,O-Dimethylhydroxylamine
hydrochloride (1.755 g, 18.0 mmol), and triethylamine (2.51 mL, 18.0 mmol) in toluene (30 mL)
at 0°C was added a solution of phosphine trichloride (0.261 mL, 3.0 mmol) in toluene (5 mL).
The reaction was then heated to 60°C for 1 h. The reaction mixture was then quenched with
saturated sodium carbonate solution and extracted with ethyl acetate. The resulting organic layer
was dried with magnesium sulfate, filtered, and concentrated under reduced pressure to give 150
mg (25%) Weinreb amide as a colorless oil.

44
LIST OF REFERENCES
1. a) Zhao, D.; Candish, L.; Paul, D.; Glorius, F. ACS Catal. 2016, 6, 5978–5988. b) Verendel, J.
J.; Paimes, O.; Dieguez, M.; Andersson, P. G. Chem. Rev. 2014, 114, 2130−2169.
2. Knowles, W. S.; Sabacky, M. J. Chem. Commun. 1968, 1445-1446.
3. Dang, T.; Kagan, H. J. Chem. Soc., Chem. Commun. 1971, 188, 481.
4. Knowles, W. S.; Sabacky, M; Vineyard, B. J. Chem. Soc., Chem. Commun. 1972, 10-11.
5. Knowles, W. S. Angew. Chem. Int. Ed. 2002, 41, 1998-2007.
6. Noyori, T.; Ito, K.; Miyashita, A; Takaya, H.; Yasuda, A; J. Am. Chem. Soc. 1980, 102, 7932-
7934.
7. Davis, M.; Crabtree, R. J. Org. Chem. 1986, 51, 2655-2661.
8. Pfaltz, A.; Lightfoot, A.; Schnider, P. Angew. Chem. Int. Ed. 1998, 37, 2897-2899.
9. Bell, S.; Wüstenberg, B.; Kaiser, S.; Menges, F.; Netscher, T.; Pfaltz, A. Science, 2006, 311,
642–644.
10. Aponick, A.; Cardoso, F. S. P.; Abboud, K. A. J. Am. Chem. Soc. 2013, 135, 14548-14551.
11. Padwa, A.; Cochran, J. E.; Kappe, C. O. J. Org. Chem., 1996, 61, 3706–3714.
12. Ochiai, K.; Takita, S.; Kojima, A.; Eiraku, T.; Iwase, K.; Kishi, T.; Ohinata, A.; Yageta, Y.;
Yasue, T.; Adams, D. R.; Kohno, Y. Bioorg Med Chem Lett 2013, 23, 375-381.
13. Corey, E. J.; Schmidt, G. Tetrahedron Lett. 1979, 20, 399-402.
14. Larsen, C. R.; Grotjahn, D. B. J. Am. Chem. Soc. 2012, 134, 10357−10360.
15. Hansen, A. L.; Ebran, J.; Gogsig, T. M.; Skrydstrup, T. J. Org. Chem., 2007, 72, 6464–6472.
16. Barton, D. H. R.; Bohe, L.; Lusinchi, X. Tetrahedron, 1990, 5273-5284.
17. Mazuela, J.; Verendel, J. J.; Coll, M.; Schaffner, B.; Borner, A.; Andersson, P. G.; Pamies, O.;
Dieguez, M. J. Am. Chem. Soc. 2009, 131, 12344–12353.
18. Takagi, R.; Igata, N.; Yamamoto, K.; Kojima, S. Journal of Molecular Catalysis A: Chemical
2010, 321, 71–76.
19. McIntyre S.; Hormann, E.; Menges, F.; Smidt, S. P.; Pfaltz, A. Adv. Synth. Catal. 2005, 347,
282 – 288.
20. Berthiol, F.; Doucet, H.; Santelli, M. Eur. J. Org. Chem. 2003, 1091-1096.

45
21. Singh, R. P.; Kamble, R. M.; Chandra, K. L.; Saravanan, P.; Singh, V. K. Tetrahedron 2001,
57, 241-247.
22. Kataoka, N.; Shelby, Q.; Stambuli, J.P.; Hartwig, J. F. J. Org. Chem., 2002, 67, 5553–5566.

46
BIOGRAPHICAL SKETCH
Bobby Owen Garrett was born in Birmingham, Alabama in 1990. He attended high
school at Corner High School in Warrior, Alabama. After high school, he attended Auburn
University on scholarship and obtained his bachelor’s degree in chemistry graduating Cum
Laude in 2013. He was an active researcher in organic chemistry at Auburn for two years under
Dr. Peter Livant and also obtained a minor in political science. He then accepted an offer to
enroll in the University of Florida Chemistry Graduate Program. There he worked on
enantioselective catalysis under Dr. Aaron Aponick where he obtained his master’s degree in
May of 2017.